Materials Studio学习笔记(一)——Materials Studio软件介绍
视频原链接:https://space.bilibili.com/677482886?spm_id_from=333.1387.follow.user_card.click
月只蓝的视频内容较为基础,适合初学者入门。
UP主页链接为: http:// https://b23.tv/TFdT21V
1 Materials Studio软件介绍
Materials Studio是由美国BIOVIA公司开发,主要用于材料的仿真和模拟,适用的范围包括原子、分子、以及宏观尺度的研究。支持量子力学、晶体预测、蒙特卡洛模拟以及介观模拟等方面。
Materials Studio软件已经在材料科学领域有多方面的应用:包括在电池领域锂离子正负极材料;在高分子材料领域包括聚合物力学性能、玻璃化转变温度(Tg)、相容性;在纳米材料领域中研究碳纳米管、石墨烯、量子点的电子与光学性质;在半导体和光电材料中应用于能带工程、载流子迁移率、激子效应;在催化材料领域应用于活性位点分析、反应路径优化等。
Materials Studio软件的界面较为友好,能够完成多尺度的模拟,涵盖了从电子结构到分子动力学,再到介观模拟的全链条研究。
2 分子模拟学
2.1 分子模拟简介
分子模拟的定义:分子性质的计算机模拟,简称为分子模拟。
分子模拟的对象:模型,即原子、分子、离子,以及它们的组合体。
分子模拟的目的:获得模型的物理、化学性质。
分子模拟的实现:量子力学、分子力学、分子动力学、耗散例子动力学‘、介观动力学。
2.2 分子模拟的尺度
分子模拟的尺度可以分为微观尺度和介观尺度。
在时间尺度上:
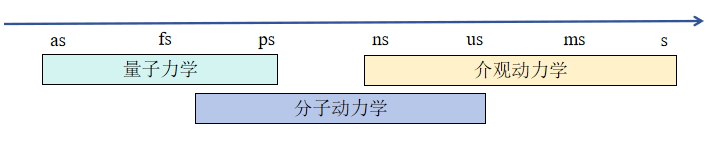
在上图中,各个符号的的意义为:
- ms:毫秒;
- us:微秒;
- ns:纳秒;
- ps:皮秒;
- fs:飞秒;
在时间尺度上,量子力学约为阿秒(秒)到皮秒(
秒)之间,分子动力学约为飞秒(
秒)到微秒(
秒)之间,介观动力学的范围为是在纳秒(
)到秒之间,
在空间尺度上:

在上图中,各个符号的意义为:
- nm:纳米;
- um:微米;
- mm:毫米;
- m:米;
在空间尺度上,纳观的典型尺度为0.1~100nm,主导的物理定律是量子力学;介观的典型尺度为100nm~100um,主导的物理定律是经典物理;微观的典型尺度为100nm~1um,主导的物理定律量子-经典过度;宏观的典型尺度大于1mm,主导的物理定律是经典力学/热力学。
2.3 经典的分子模拟过程
经典的分子模拟过程可以分为如下几步:
- 构建模型
- 设计计算参数
- 运行计算
- 分析和展示计算结果
完成了上述四个步骤,既可以完成一次分子模拟过程。
2.4 选择模型
选择模型的过程是分子模拟中重要的过程,直接影响模拟结果。
2.4.1 更具自己研究的需求、期望获得的结果
Materials Studio模块的类型,可以分为5个大类:量子力学模块、分子力学模块、蒙特卡洛模块、粗粒化或介观类模块。每个模块都有其他模块没有的、特定的功能。例如:
- 能带、态密度->量子力学类的模块:CASTEP、
。
- 经典势图-> 量子力学的模块:
- 晶体形貌->晶体学模块:Morphology。
- 结合能、吸附能->
- 力学性质、弹性常数->
2.4.2 根据自己构建模型的尺寸,原子数来构建模型
当模型较小时,可以使用量子力学模块(数十个或者数百个例子);当模块中等大小,包含数千数万个原子,可以使用分子力学的模块;当模块较大,包含大量的原子(数万个乃至更多)可选择结合能、介管模型类模块。
2.4.3 根据模拟的过程是否涉及到电子的得失和转移
涉及电子的得失和转移的过程,例如化学机理研究、过渡态搜索,应当选择量子力学的模块。
分子力学理论本身构建的思想,就是要舍弃那些“数目庞大、行为复杂,却微不足道”的电子,所以从基本理论上,就决定了分子力学类的模块,不具备考察电子行为的能力。
2.4.4 根据模拟对立场的要求
分子类模型的计算速度较快,而且计算精度尚可,应用非常广泛。
但是,分子力学模块有很大的局限性,就是力场,更确切地说,是力场的适用性,力场是分子力学计算的基础,模型中任何一个原子力场参数的缺失,都会导致计算失败。遗憾的是,目前还没有哪个力场,其适应性足以涵盖绝大多数原子、离子、官能团、粒子团。
如果找遍了各类力场,还是无法获得对自己研究的模型具有完整适应性的力场,此时可以考虑转向量子力学类的模块。
2.4.5 根据已有的文献报告
当研究与现有文献相似时,借鉴文献作者对于MS模块的选用结果。官方网站为MS官方网站,认识MS发挥MS的优势,高度继承性实现多模块协作。