【文献阅读】建立高可信度的阴性样本,改进化合物-蛋白质相互作用预测
文章链接:Improving compound–protein interaction prediction by building up highly credible negative samples | Bioinformatics | Oxford Academic

动机:化合物-蛋白质相互作用(CPIs)的计算预测对于药物设计和开发非常重要,因为CPIs的基因组规模实验验证不仅耗时而且昂贵。随着越来越多的可验证交互的可用性,由于缺乏可靠的负CPI样本,计算预测方法的性能受到严重影响。一种系统的筛选可靠的阴性样本的方法对于提高计算机预测方法的性能至关重要。
补充材料获得: Supplementary files.
研究背景
- 研究问题:这篇文章主要研究如何通过构建高度可信的负样本,来改进化合物-蛋白质相互作用(CPIs)的预测。由于基因组规模的CPIs实验验证既耗时又昂贵,因此计算预测方法在这一领域具有重要意义。
- 研究难点:该问题的研究难点在于缺乏可靠的负样本。大多数现有的计算模型假设相似的化合物可能与相似的目标蛋白质相互作用,但这些模型在缺乏可靠负样本的情况下表现不佳。
- 相关工作:传统的计算方法分为基于结构和基于配体的两类,但都存在一定的局限性。近年来,基于机器学习的方法通过整合化学属性、基因组属性和已知的CPIs取得了显著成功。然而,这些方法仍然受到已知相互作用的限制,导致预测失败。
研究方法
这篇论文提出了一种通过逆否命题来筛选高度可信的负样本的方法。具体来说,假设一个蛋白质与给定化合物的所有已知或预测靶点都不相似,则该化合物不太可能与该蛋白质相互作用。研究方法包括以下几个步骤:
- 数据整合:整合了多种化合物和蛋白质的数据资源,包括化学结构、化学表达谱、化合物的副作用、蛋白质的氨基酸序列、蛋白质-蛋白质相互作用网络和功能注释。
- 相似性计算:计算化合物和蛋白质的多维度相似性,并将其整合为单一的综合相似性度量。公式如下:
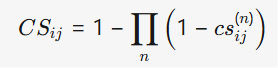
其中,csij(n)cs_{ij}^{(n)}csij(n)表示从化学结构和副作用中得出的相似性度量。蛋白质的综合相似性计算公式类似:
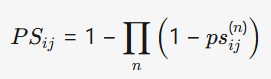
其中,psij(n)表示从序列相似性、功能注释语义相似性和蛋白质域相似性中得出的相似性度量。
3. 负样本筛选框架:基于逆否命题,假设一个蛋白质与给定化合物的所有已知或预测靶点都不相似,则该化合物不太可能与该蛋白质相互作用。具体步骤包括计算候选负样本的距离得分,并根据得分排序和过滤。
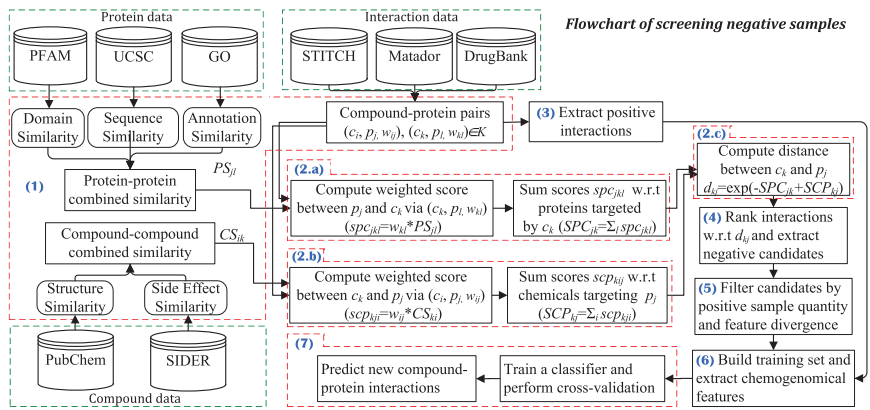
Fig.1.The flowchart of our negative CPI screening framework.Three green dashed-line boxes show the data resources used in our screening process, and the red dashed-line boxes represent the screening steps that include multiple operations.
实验设计
- 数据收集:从DrugBank、Matador和STITCH数据库中检索CPIs数据。选择了2290630个人类CPIs和2141740个秀丽隐杆线虫CPIs。
- 化学数据:从PubChem数据库获取药物的化学结构指纹,并计算Jaccard分数作为化学结构相似性。从SIDER数据库获取药物的副作用,并计算Jaccard分数作为副作用相似性。
- 蛋白质数据:从UCSC Table Browser获取蛋白质的氨基酸序列,并计算归一化的Smith-Waterman分数作为序列相似性。从GO数据库获取蛋白质的功能注释,并计算Jaccard分数作为功能注释语义相似性。从PFAM数据库提取蛋白质域,并计算Jaccard分数作为蛋白质域相似性。
- 交叉验证:采用成对交叉验证和块交叉验证两种协议来评估负样本的性能。成对交叉验证假设目标是检测已知配体化合物和已知靶蛋白之间的缺失相互作用,而块交叉验证假设目标是检测新配体化合物和新靶蛋白之间的新相互作用。
结果与分析
1. 经典分类器性能评估:在人类和小鼠数据集上,使用六种经典分类器(朴素贝叶斯、kNN、随机森林、L1-logistic回归、L2-logistic回归和支持向量机)进行性能评估。结果显示,所有分类器在使用筛选出的负样本时,性能显著优于随机生成的负样本。
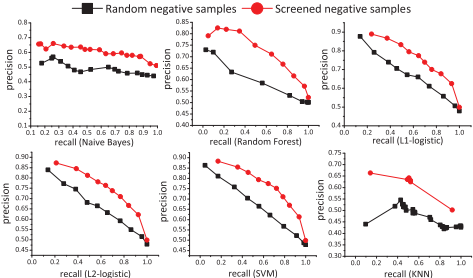
Fig.3. Precision-recall curves of six classical classifiers on screened and ran-domly generated negatives of human(blockwise cross-validation)
2. 现有预测方法评估:评估了三种现有的预测方法(BLM、GIP核和KBMF2K)在使用筛选出的负样本时的性能。结果显示,所有方法在使用筛选出的负样本时,性能显著提升。
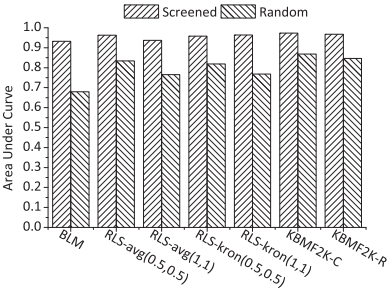
Fig. 4. Histogram of the AUC values achieved by three existing predictive methods on screened and randomly generated negative samples of human
3. 药物生物活性数据集验证:使用激酶抑制剂的定量药物-靶标生物活性测定数据集进行验证。结果显示,使用筛选出的负样本训练的支持向量机和L2-logistic分类器在精度-召回率曲线上表现优异。
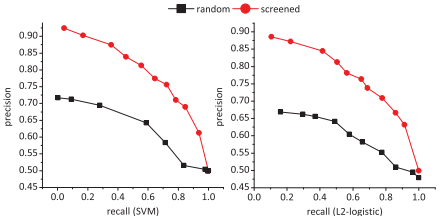
Fig.5. Precision-recall curves of the SVM and L2-logistic classifiers trained on screened and randomly generated negative samples, evaluated on the kinase bioactivity assay data(Davis et al.,2011)
4. 新相互作用预测:基于筛选出的负样本和DrugBank中的正样本,训练支持向量机分类器预测新的CPIs。结果显示,预测的新相互作用在功能和疾病相关注释中高度富集。
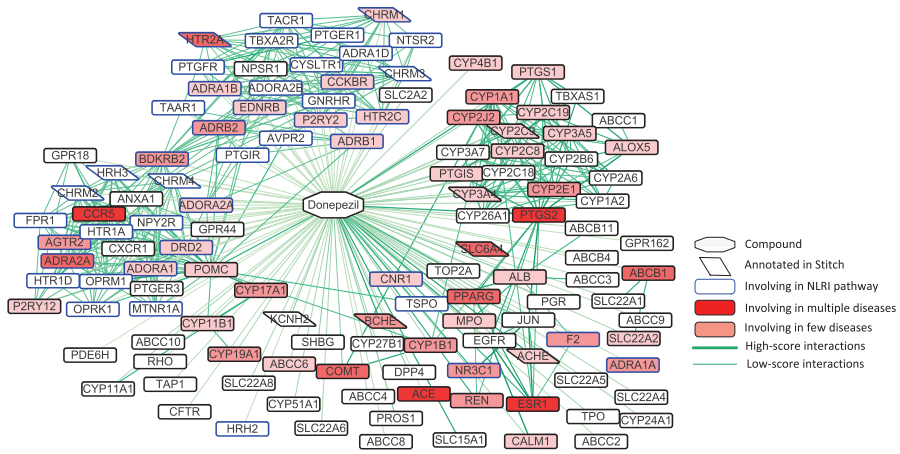
Fig.6. Predicted target proteins of Donepezil and related functional annotations, including neuroactive ligand-receptor interaction pathways, diseases recorded in DrugBank and STITCH
总体结论
这篇论文提出了一种系统的方法来筛选高度可信的负样本,显著提升了现有计算方法的性能。通过整合多种数据资源和创新的筛选框架,研究不仅提供了高质量的负样本,还为药物靶点发现和现有数据库的补充提供了有价值的资源。