ThermoMPNN/ThermoMPNN-D 的安装及使用教程
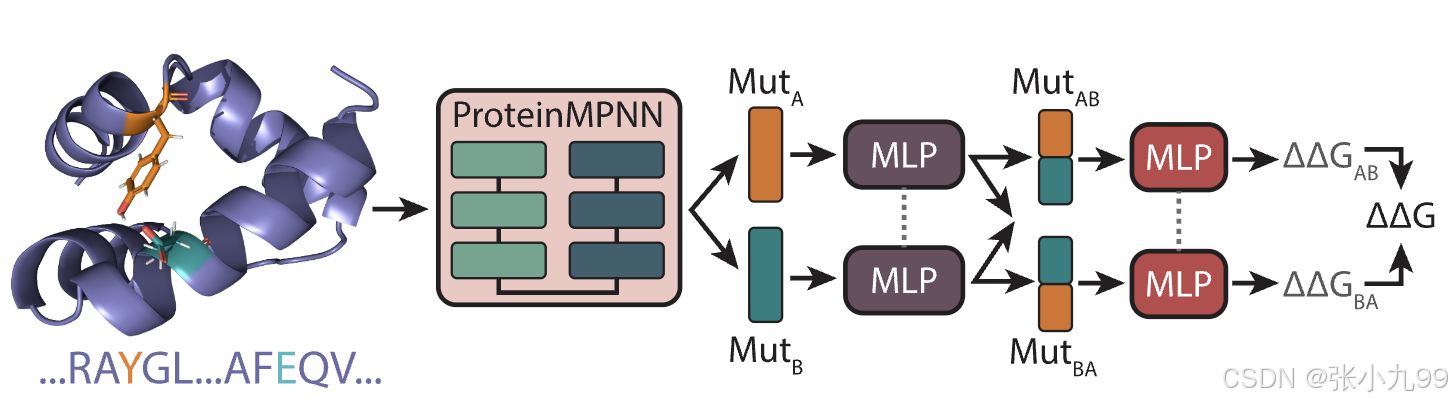
写在前面:ProteinMPNN使用了整个PDB中的19700个蛋白簇进行训练,目标是根据给定的蛋白质骨架预测天然序列,它通过预测每个位置的天然残基的概率来实现这一点。这些预测是基于从PDB中的天然蛋白质中学习到的结构模式。知道了ProteinMPNN的原理,就引出了ThermoMPNN的动机:通过使用预训练的ProteinMPNN嵌入作为输入特征,结合迁移学习,利用了序列恢复和稳定性优化任务之间的知识重叠:已知结构的天然蛋白质序列通常被认为在常温下至少是边际稳定的,因此,ProteinMPNN预测的氨基酸概率与相应点突变体的稳定性变化应具有相关性。
一、安装教程
1、克隆仓库到本地
git clone https://github.com/Kuhlman-Lab/ThermoMPNN-D.git
cd ThermoMPNN-D2、安装在 environment.yaml 中准备好的 python 依赖项(建议使用 mamba)
mamba env create -f environment.yaml # 这里也可以使用conda安装 conda env create -f environment.yaml
3、将 ThermoMPNN 添加到您的 PYTHONPATH,以便 python 可以找到所有模块
export PYTHONPATH=$PYTHONPATH:/path/to/ThermoMPNN-D4、问题一:cuda 报错问题 undefined symbol: cudaGraphInstantiateWithFlags, version libcudart.so.11.0
(1)问题原因:版本冲突
PyTorch 版本:
2.0.1是为 CUDA 11.7 编译的。系统 CUDA 工具包:
nvcc显示 CUDA 11.5(实际用于编译的版本)。驱动支持:
nvidia-smi显示驱动支持 CUDA 12.2(兼容性高,但需工具包匹配)。(2)关键矛盾:PyTorch 依赖的 CUDA 运行时(如
libcudart.so.11.7)与系统中的 CUDA 工具包(11.5)不匹配。(3)解决方案:在 Conda 环境中安装 CUDA 11.7 工具包
首先,激活当前环境
conda activate ThermoMPNN-D其次,安装 CUDA 11.7 工具包
conda install cudatoolkit=11.7 -c pytorch最后,验证CUDA可用性,输入为:True,则表明安装成功
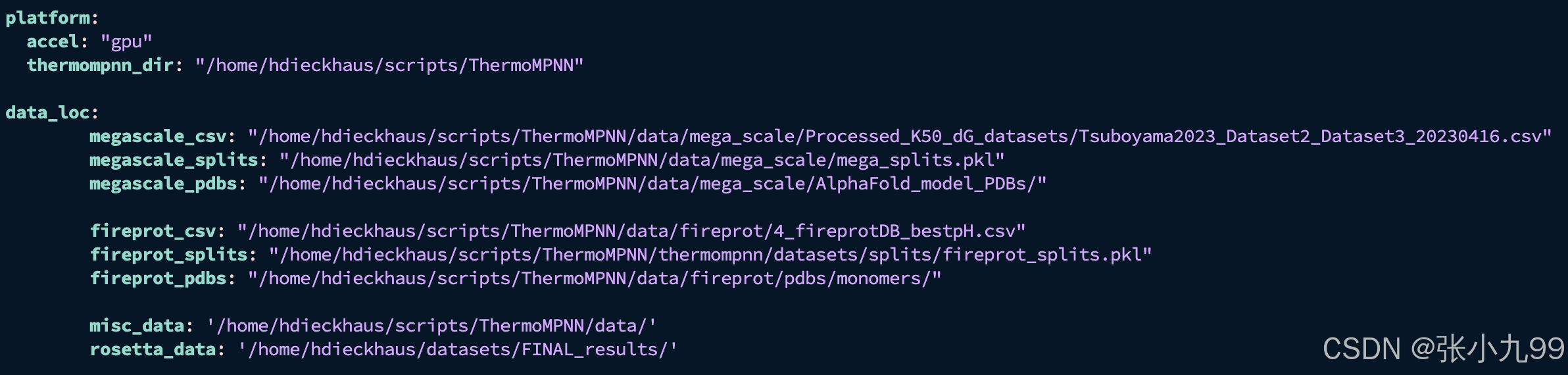
python -c "import torch; print(torch.cuda.is_available())"5、最后,修改 examples/configs/local.yaml 中的本地文件路径信息以匹配您的系统
vim examples/configs/local.yaml把该文件中的路径修改为自己下载的文件路径, 如果主要是用该模型进行推理的话,那么实测秩序要修改thermompnn_dir的路径为自己git clone的文件路径即可;
6、问题二: RuntimeError: Numpy is not available。执行示例脚本的时候可能还会有一个错误,原因是当前使用的 NumPy 是 2.x 版本,而某些依赖(如 PyTorch、torchmetrics、pybind11 编译的模块等)是基于 NumPy 1.x 编译的,导致兼容性崩溃或 NumPy 根本无法正常初始化使用。
解决方法:降级 NumPy 到 <2.0 版本,即可正常运行!
pip install "numpy<2.0"
注意:安装好虚拟环境之后,ThermoMPNN和ThermoMPNN-D两个仓库是可以共用一个虚拟环境的,所以安装ThermoMPNN只需要执行以下指令下载并修改local.yam文件中的项目路径即可,如果不重新训练模型的话,只需要修改thermompnn_dir的路径即可!
git clone https://github.com/Kuhlman-Lab/ThermoMPNN.git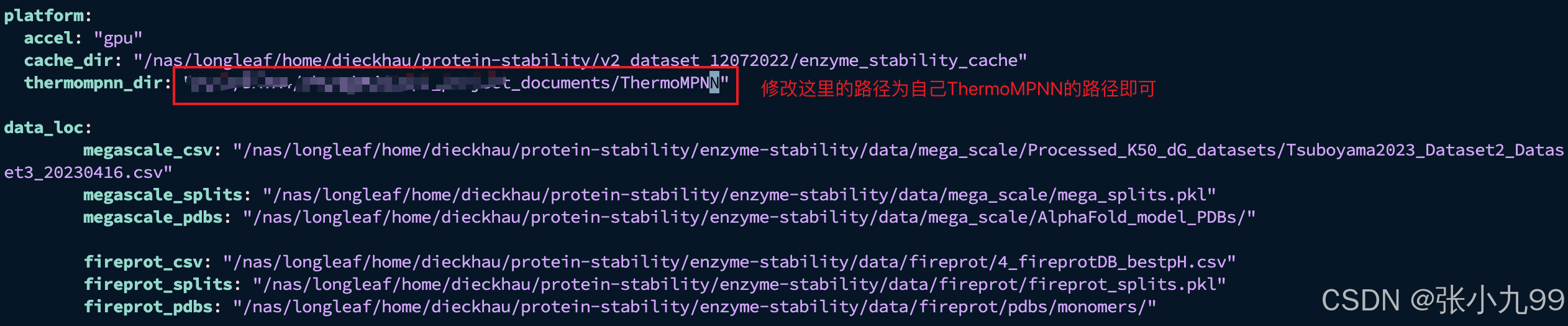
二、使用教程
(一)ThermoMPNN使用教程
1、将以下文件保存为 inference.sh 文件;
repo_location="/ThermoMPNN/analysis"cd $repo_locationpython custom_inference.py \--pdb ../1_project/test/AF-P31456-F1-model_v4.pdb \--chain A \--model_path ../models/thermoMPNN_default.pt \--out_dir ../1_project/test/2、将该脚本修改为可执行文件;
chmod +x inference.sh3、执行以下脚本去运行ThermoMPNN;
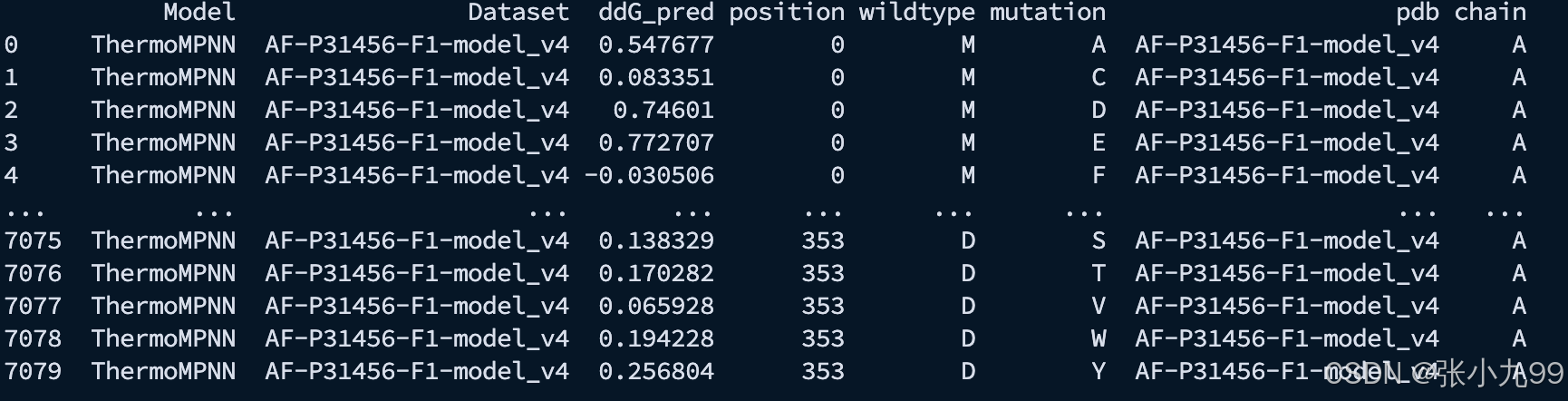
./inference.sh4、运行完成之后,输出结果示例如下;
(二)ThermoMPNN-D使用教程
1、单突变模型(Single Mutant Model)
功能:这是一个更新版的单突变 ThermoMPNN 模型。该模型使用了更少的参数,并且进行了适当的批量推理,以提高预测速度。预测结果与之前发布的 ThermoMPNN 模型相似。
使用方法:通过以下命令运行该模型:
python v2_ssm.py --mode single --pdb 1VII.pdb --batch_size 256 --out 1VII
2、加性双突变模型(Additive Double Mutant Model)
功能:这个模型通过将每个单一突变的贡献相加来生成双突变模型的预测结果,而不考虑表观遗传效应(即不考虑突变之间的相互作用)。与表观遗传模型相比,这种方法推理速度更快,因为它仅仅是将单一突变的贡献加在一起,而不需要分别预测每个突变。
使用方法:通过以下命令运行该模型:
python v2_ssm.py --mode additive --pdb 1VII.pdb --batch_size 256 --out 1VII
3、上位双突变模型(Epistatic Double Mutant Model)
功能:该模型试图捕捉双突变之间的表观遗传相互作用(即突变之间的相互影响)。由于要对每个单独的突变进行推理,推理速度比单一突变模型和加性模型慢一些。但通过一些向量化和批量化的技巧,推理时间仍然是比较快的(通常少于 1 分钟)。
使用方法:通过以下命令运行该模型:
python v2_ssm.py --mode epistatic --pdb examples/pdbs/1VII.pdb --batch_size 2048 --out 1VII
关键参数说明
| 参数 | 作用 |
|---|---|
--mode | 指定模型类型(single/additive/epistatic) |
--pdb | 输入蛋白质结构文件(PDB 格式)路径 |
--batch_size | 批量大小,值越大速度越快(受 GPU 显存限制) |
--out | 输出结果目录前缀(如 1VII 会生成 1VII_single.csv 等文件) |
(1)--threshold 参数
-
作用:
--threshold参数控制哪些突变将被保存到磁盘上。它主要影响的是稳定性变异(stabilizing mutations)和不稳定性变异(destabilizing mutations)的筛选。 -
默认设置:默认情况下,ThermoMPNN 只会保存 稳定性突变,即那些ddG <= -0.5 kcal/mol 的突变。这里的 ddG 表示突变后的自由能变化(ΔG),值为负表示突变带来了稳定化的效果。
-
如何影响:
-
ddG <= -0.5 kcal/mol 的突变被认为是稳定化突变,这些突变使得蛋白质结构更稳定。
-
其他突变(例如 destabilizing mutations,稳定性差的突变)默认不会保存到磁盘。
-
-
自定义设置:
-
如果想要保存 所有的突变(包括稳定性差的突变),可以将
--threshold设置为非常高的值(例如,--threshold 100),这样可以确保所有的突变都会被保存。
-
(2)--distance 参数
-
作用:
--distance参数是用于加性模型(additive model)或上位模型(epistatic model)中的,决定了过滤突变时的“距离阈值”。 -
目的:该参数用于筛选在空间上“接近”的氨基酸残基,认为这些残基更可能具有上位相互作用(两个突变的联合效应不等于各自单独效应的简单相加(如突变 A 的效果可能依赖突变 B 的存在))。
-
如何工作:
-
假设空间上邻近的残基更可能发生上位相互作用,因此通过
--distance限制计算范围,平衡精度与速度。 -
较小的
--distance值(例如,2 Å 或 3 Å)会导致 更严格的筛选,只考虑那些非常接近的残基对。这样做有助于在推理时聚焦于可能相互作用的氨基酸对。 -
默认值是 5 Å,基于 Ca-Ca(α-碳到α-碳的距离) 的距离。如果两个氨基酸残基之间的 Ca-Ca 距离小于 5 Å,则它们被认为是“接近”的,可能存在上位相互作用。
-
总结:ProteinMPNN本质上是通过大量数据学习了氨基酸序列与结构之间的关系它学到的不仅仅是"最优"序列,而是在给定结构下的氨基酸概率分布。这个概率分布反映了序列-结构适配度,某种程度上可以类比为自由能景观。打分差异反映了局部环境适配度的变化,与实验测定的ΔΔG有物理对应关系。这种方法可能难以准确预测一些特殊情况:涉及远程相互作用的突变、引起构象大变化的突变、多重突变的协同效应。总的来说,这种方法之所以能work,是其捕捉了序列-结构-稳定性之间的复杂关系。
参考链接:
1、https://github.com/Kuhlman-Lab/ThermoMPNN
2、https://github.com/Kuhlman-Lab/ThermoMPNN-D
3、PNAS | ThermoMPNN:基于ProteinMPNN的蛋白质热稳定性预测