利用参考基因组fa和注释文件gff提取蛋白编码序列
目录
gffread提取
提取名称对应表格
替换转录本名称为基因名
提取最长转录本获取编码序列
一个提醒
其实提取蛋白序列是生信分析中相当常见的一个操作,但是我在回顾和查找各类提取方法时,踩了不少坑,故在此记录一下本人的提取全流程方法。
事情的起因是我需要在某一步软件的准备阶段进行蛋白序列的BLAST比对,需要一个以基因名称命名蛋白序列的文件,于是查找我的基因组gff文件如下
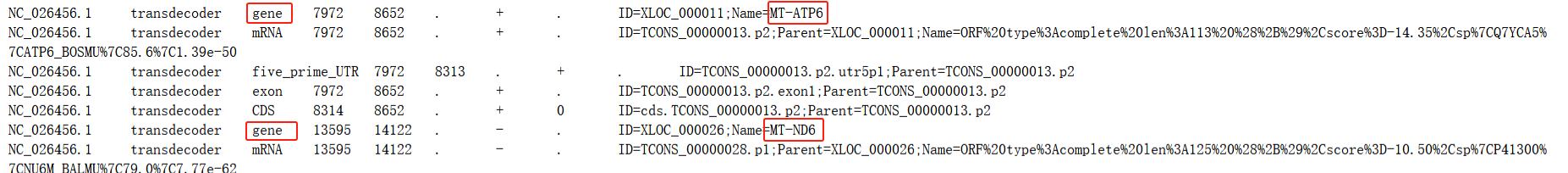
这里我需要的其实是gene这一行的所有序列,并使用“Name=”之后的基因名称命名这段序列
gffread提取
一番查找后,我首先尝试了最基础的gffread
gffread -g genome.fasta -y protein.fa genome.gff3理想状态下,使用了-y参数即可提取我们需要的蛋白编码序列protein.fa,这里genome.fasta是参考基因组序列,genome.gff3是对应的参考注释文件
但是检查结果发现这里提取出来的其实是注释文件中mRNA这一行的所有对应序列片段,且命名方式是“ID=”之后的转录本编号,而不是我们需要的“Name=”之后的基因名称
提取名称对应表格
于是我决定先将所有的转录本编号先替换成对应的基因名称,即我们需要一个gene ID + gene Name + mRNA ID三个字段的对应表格,将目前mRNA ID替换成对应的gene Name,一个基因可能会对应多个片段的mRNA
提取对应名称的bash脚本如下,结果文件为 mRNA_mapping.txt
#!/bin/bash# 输入文件路径列表(以ABC三个物种为例)
input_gffs=("A.gff3""B.gff3""C.gff3"
)
# 输出目录(可替换成自己的路径)
output_dir="/data/work/out"
# 确保输出目录存在
mkdir -p "$output_dir"
# 遍历每个 GFF 文件并生成对应的表格
for input_gff in "${input_gffs[@]}"; do# 获取文件名(不包含路径)filename=$(basename "$input_gff")# 根据文件名设置输出表格路径case "$filename" in"A.gff3")output_table="$output_dir/A_mRNA_mapping.txt";;"B.gff3")output_table="$output_dir/B_gene_mRNA_mapping.txt";;"C.gff3")output_table="$output_dir/C_gene_mRNA_mapping.txt";;*)echo "未知的文件名: $filename"continue;;esac# 使用 awk 提取所需信息并生成表格awk -F'\t' 'BEGIN {OFS="\t"; print "geneID", "name", "mRNA"} {if ($3 == "gene") {# 提取 gene 的 ID 和 Name 字段match($9, /ID=([^;]*)/, arr);gene_id = arr[1];match($9, /Name=([^;]*)/, arr);gene_name = arr[1];# 保存到 gene_map 中gene_map[gene_id] = gene_name;} else if ($3 == "mRNA") {# 提取 mRNA 的 ID 和 Parent 字段match($9, /ID=([^;]*)/, arr);mRNA_id = arr[1];match($9, /Parent=([^;]*)/, arr);parent_id = arr[1];# 如果 Parent ID 在 gene_map 中存在,则输出对应的 geneID、name 和 mRNAif (parent_id in gene_map) {print parent_id, gene_map[parent_id], mRNA_id;}}}' "$input_gff" > "$output_table"echo "表格生成完成,输出文件为:$output_table"
done生成的表格格式如下,我们需要的就是将第三列的mRNA名称替换成第二列的基因name
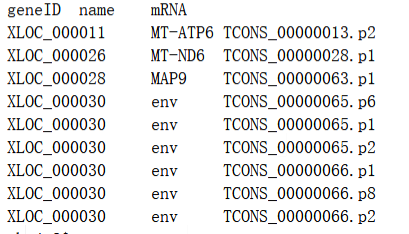
替换转录本名称为基因名
于是利用上一步生成的表格,我们进行名称的替换,生成新的重命名蛋白序列
替换对应名称的python脚本如下,得到 protein_renamed.fa
#!/usr/bin/env python3import sys
import os# 输入文件路径列表
mapping_files = ["A_gene_mRNA_mapping.txt","B_gene_mRNA_mapping.txt","C_gene_mRNA_mapping.txt"
]protein_files = ["A_protein.fa","B_protein.fa","C_protein.fa"
]
# 可以替换成自己的输出路径
output_dir = "/data/work/out"# 确保输出目录存在
os.makedirs(output_dir, exist_ok=True)# 遍历每个映射表和蛋白文件
for mapping_file, protein_file in zip(mapping_files, protein_files):# 读取映射表,生成替换规则mRNA_to_name = {}with open(mapping_file, "r") as infile:next(infile) # 跳过表头for line in infile:gene_id, name, mRNA = line.strip().split("\t")mRNA_to_name[mRNA] = name# 设置输出文件路径output_file = os.path.join(output_dir, os.path.basename(protein_file).replace("protein.fa", "protein_renamed.fa"))# 替换序列名称with open(protein_file, "r") as infile, open(output_file, "w") as outfile:for line in infile:if line.startswith(">"):# 提取当前序列名称current_id = line.strip().lstrip(">")# 如果当前序列名称在映射表中,替换为对应的 Nameif current_id in mRNA_to_name:new_name = mRNA_to_name[current_id]outfile.write(f">{new_name}\n")else:outfile.write(line) # 如果没有找到映射,保留原名称else:outfile.write(line) # 序列行直接写入输出文件print(f"序列名称替换完成,输出文件为:{output_file}")提取最长转录本获取编码序列
替换完基因名称后,我们需要解决的就是重复的转录本问题,一般来讲在后续的分析中,每个编码蛋白对应的序列只需要取最长的那条转录本即可,而我们提取的序列文件中,一个蛋白明显存在许多重复的转录本(如截图中的env)
于是我们需要再写一个脚本提取同一个基因名称下最长的那条转录本翻译的蛋白序列,才是我们需要的真正的蛋白编码序列 protein_longest.fa
这里我借用了一个简单的python包,下载很方便,pip install即可,详细用法感兴趣可见第1章 介绍 — Biopython-cn 0.1 文档
from Bio import SeqIO
from Bio.SeqRecord import SeqRecorddef extract_longest_transcripts(input_fasta, output_fasta):"""从输入的 FASTA 文件中提取每个基因的最长转录本,并保存到输出文件中。:param input_fasta: 输入的 FASTA 文件路径:param output_fasta: 输出的 FASTA 文件路径"""# 读取 FASTA 文件,存储每个基因的所有转录本gene_transcripts = {}for record in SeqIO.parse(input_fasta, "fasta"):gene_name = record.id # 假设序列 ID 就是基因名if gene_name not in gene_transcripts:gene_transcripts[gene_name] = []gene_transcripts[gene_name].append(record)# 筛选每个基因的最长转录本longest_transcripts = []for gene_name, transcripts in gene_transcripts.items():# 按序列长度降序排序,取最长的转录本longest_transcript = max(transcripts, key=lambda x: len(x.seq))longest_transcripts.append(longest_transcript)# 将最长转录本保存到新的 FASTA 文件with open(output_fasta, "w") as output_handle:SeqIO.write(longest_transcripts, output_handle, "fasta")print(f"Longest transcripts saved to {output_fasta}")# 输入文件列表
input_files = ["A_protein_renamed.fa","B_protein_renamed.fa","C_protein_renamed.fa"
]# 对每个输入文件处理并生成对应的输出文件
for input_fasta in input_files:# 生成输出文件路径,通过替换文件名中的 "renamed" 为 "longest" 来生成输出文件名output_fasta = input_fasta.replace("renamed", "longest")extract_longest_transcripts(input_fasta, output_fasta)最后less检查一下,此时重复的env基因已经被去除了,每个基因名称下对应的都是最长的那条转录本的蛋白翻译序列,大功告成!

一个提醒
之所以说提取过程中踩了很多坑,一个是由于本身gff注释文件的手动修改,还有一个就是各类古早软件的默认参数并不是像字面上那么精准,能够提取到我们想要的东西,很多时候真的要根据先验知识一步步检查中间文件是否合理
在发现biopython这个包时,我发现它也自带一个类似的蛋白编码提取的函数,代码如下
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord# 假设GFF文件和参考基因组文件已经准备好
gff_file = "A.gff3"
fasta_file = "A.fasta"# 解析GFF文件,提取所有第三列为“gene”的条目
genes = []
with open(gff_file, "r") as gff:for line in gff:if line.startswith("#"):continuefields = line.strip().split("\t")if fields[2] == "gene":chrom, start, end, strand = fields[0], int(fields[3]), int(fields[4]), fields[6]name = fields[8].split(";")[1].split("=")[1]genes.append((chrom, start, end, strand, name))# 提取基因序列并翻译为蛋白序列
with open(fasta_file, "r") as fasta:genome = SeqIO.to_dict(SeqIO.parse(fasta, "fasta"))# 创建一个列表存储蛋白序列
protein_sequences = []for chrom, start, end, strand, name in genes:if chrom in genome:seq = genome[chrom].seq[start-1:end] # 提取序列if strand == "-":seq = seq.reverse_complement() # 如果是负链,反向互补protein_seq = seq.translate() # 翻译为蛋白序列protein_sequences.append(SeqRecord(protein_seq, id=name, description=f"{chrom}:{start}-{end}"))# 将蛋白序列保存到文件
with open("/data/work/out/A_protein.fa", "w") as output:SeqIO.write(protein_sequences, output, "fasta")这里根据教程我们提取了A_protein.fa,看似过程很合理,提取了注释文件中标记为“gene”的一行,这一行其实对应的也就是最长的转录本,但是检查结果时发现格式如下

不仅多了很多代表终止密码子的*,而且对比之前的编码蛋白序列还多了不少密码子,检索后发现是提取了对应的非编码区(如 5' UTR 和 3' UTR),其实只需要CDS
但是仔细修改一下细节这个方法也是相当可行的,这里仅作提醒hh
希望大家做的时候不要踩类似的坑,比如 带5' UTR 和 3' UTR的蛋白序列其实要大不少