论文学习:《EVlncRNA-net:一种双通道深度学习方法,用于对实验验证的lncRNA进行准确预测》
原文标题:EVlncRNA-net: A dual-channel deep learning approach for accurate prediction of experimentally validated lncRNAs
原文链接:https://www.sciencedirect.com/science/article/pii/S0141813025020896
长链非编码RNA( long non-coding RNAs,lncRNAs )指的是长于200核苷酸的不编码蛋白质的转录物。lncRNA在许多生物学过程中发挥重要作用,包括基因调控、发育和免疫应答。此外,lncRNAs还与多种人类疾病有关,如癌症、肌肉疾病和遗传性疾病。
高通量测序技术(High-throughput sequencing)又称“下一代”测序技术(Next-generation sequencing technology),能够一次并行对大量核酸分子进行平行序列测定的技术。
低通量测序技术(Low-throughput sequencing)指的是较小规模的DNA测序技术。
本文提出了一种新的基于序列语言处理的深度学习框架实验验证lncRNAs网络( EVlncRNAs-net )。该框架包含两个表示学习模块:EVlncRNA-net ( GCN )和EVlncRNA-net ( CNN )。GCN 引入了一种新颖的图构造方法和专门的节点编码技术,该模块将lncRNA序列转化为图形格式,并使用图卷积进行处理。CNN 通过卷积神经网络从one-hot编码序列中提取特征。
面对的问题:高通量测序中鉴定出的lncRNA数量与实验验证的lncRNA数量之间的差距。
LncADeep使用深度信念网络整合内在和同源特征进行lncRNA识别,同时通过基于序列和结构的深度神经网络预测lncRNA-蛋白质相互作用。
LncRNA _ Mdeep是一种非比对、多模态深度学习方法来区分lncRNA和蛋白质编码转录本,将多模态输出连接到一个深度神经网络进行最终分类。
EVlncRNA-pred,一种基于SVM的方法,旨在区分实验验证的lncRNAs ( EVlncRNAs )和高通量测序RNAs ( HTlncRNAs )。
EVlncRNA-Dpred将k-mer特征融入深度神经网络( DNN ),one-hot编码融入卷积神经网络( CNN )。但较大的k-mer值会导致编码向量稀疏且质量较低。
为了克服这些局限性,本文开发了一种新型的双模块EVlncRNA-net用于EVlncRNA的预测。EVlncRNA-net结合了图卷积网络( GCN )和卷积神经网络(CNN )进行综合表示提取。
1.GCN提出了一种新颖的图构建方法和专门的节点编码技术,该模块将lncRNA序列转换为图结构,基于3-mer多尺度区间信息和编码构建节点,然后利用GCN从这些图中提取表示。有效解决了传统kmer编码忽略序列细节的局限性,避免了与传统编码方法相关的稀疏性和质量损失。
2.CNN模块将序列转化为one-hot编码数据,并使用CNN和线性层提取深层特征。
3.模型整合两个模块的特征,并通过全连接层产生最终的预测。
EvlncRNA-Net概述
EVlncRNA-net的基本架构如图1所示。输入为lncRNA或ncRNA序列,输出为该序列被预测为EVLncRNA的概率。该过程的核心在于表示学习,它包括两个主要模块:GCN和CNN。在GCN模块中,lncRNA使用k-mer序列片段来描述,称为" k-mer单元"。每个k-mer单元由4的k次个初级特征节点和单个k-mer特征节点组成。为了平衡节点维度和生物相关性,我们为我们的k-mer单元选择一个k值为3。主要原因是3-mers由于其在保持维度可管理的同时有效地表示序列而被广泛应用于生物信息学中。此外,3-mers提供了最优的特征维度。较大的k-mer规模会导致主要特征节点的数量呈指数增长,显著提高复杂度。
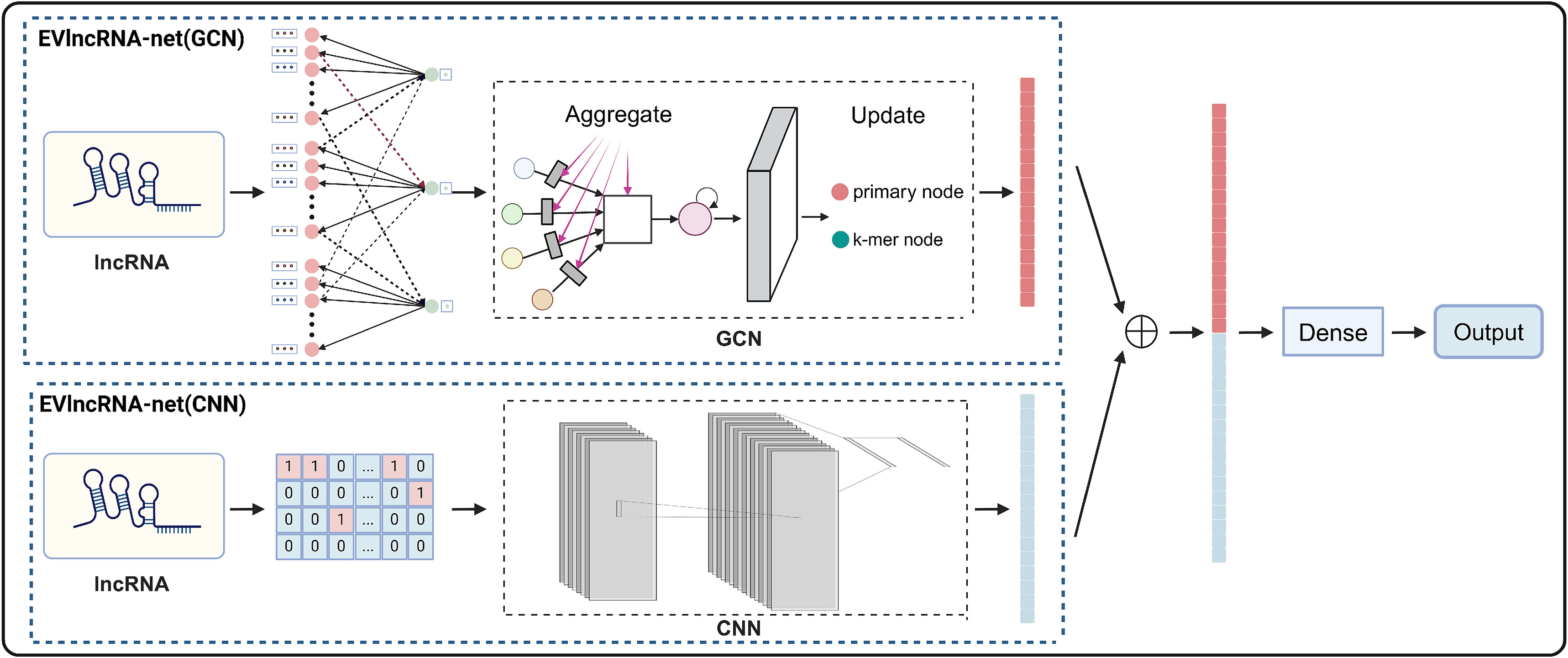
对于每个3-mer单元,64个主要节点分别代表当前3-mer和64种3-mer之间的连接。这些节点的特征值来自于多尺度区间连接。k-mer特征节点表示特定3-mer单位在序列中出现的频率。
在将lncRNA序列转换为图结构后,GCN模块使用三个SAGEConv图卷积层来细化节点特征。在每一层中,主特征节点根据其连接的k-mer特征节点的信息进行更新,进而增强k-mer特征节点。在此优化之后,模型使用带有线性层的全局图池化方法聚合了所有节点的主要特征,产生了捕获整个图的特征表示,进而准确地表示了原始序列。这种设计使得模型可以有效地处理非欧几里得数据。通过将不同长度的lncRNA转化为图结构,该模型克服了通常与传统序列比对和分析方法相关的挑战。通过利用图卷积的灵活性和抽象性,GCN展示了其捕获复杂序列模式的能力,提高了信息集成,并保持了自适应性。
在CNN模块中,首先将lncRNA序列转换为one-hot编码。这些编码后的特征再经过两个连续的卷积层。随后,批量归一化层和ReLU激活函数进一步细化了特征。然后,该模型应用自适应最大池化层和线性层来降低池化输出的维度。该序列操作使CNN模块能够从lncRNA中提取深层特征表示,这对于识别序列内的局部模式和提高分类精度至关重要。
在得到两个模块的特征后,将它们进行组合,并通过一个线性层产生最终的分类概率。
GCN特征提取
图构建

(A)将序列转换为图形形式的一般过程。
(B)将序列划分为3-mer片段,每个3-mer构成一个3-mer单位,它是图的一个基本单位。每个3-mer单元由64个初级特征节点和1个k-mer特征节点组成,代表两种不同类型的节点。
(C) 3-mer单元内的特征提取步骤。
(D)完全图中两类节点之间的最终连接关系。每个3-mer单元的节点通过特定的关系连接起来,形成lncRNA序列的最终图表示
将序列划分为3-mer单元后,下一步的工作是在每个单元之间建立节点连接。3-mer单元之间的连接是通过连接整个图中的k-mer特征节点和主要特征节点来实现的。每个3-mer单元中的k-mer特征节点连接其64个主要特征节点。这些连接的方向是从k-mer特征节点到主要特征节点,代表从该特定3-mer开始的所有3-mer对。同样,初级特征节点在其对应的对中也连接到后续3-mer的k-mer特征节点。
这个过程导致了序列的图形表示,如图2(D)所示,用实线表示从k-mer节点到主节点的连接,虚线表示从主节点到k-mer节点的连接。这些节点连接关系影响着后续连接节点之间的相互更新。
对k-mer特征节点进行k-mer编码
k-mer是由核苷酸序列中的K个连续核苷酸组成的子串。在一个RNA序列中,核苷酸由A、U、C和G表示,产生4^k个不同的k-mers单位。对于K = 3,有64个不同的3-mer单位,包括AAA,AAC,AAU,AAG,..,GGG,共64个不同的3-mer单位。在GCN模块中,3-mer单元构成了lncRNA的基本组成部分。每个3-mer单元包括64个主节点和1个k-mer特征节点。将3-mer的频率作为k-mer特征节点的特征值。
对主要特征节点进行编码
主特征节点是每个3-mer单元的重要组成部分。每个3-mer单元包含64个初级特征节点。图对主要特征节点进行编码,用邻接矩阵表示,邻接矩阵捕捉3-mers之间在指定间隔d的连接。该邻接矩阵被用于编码基于多尺度间隔d的3-mer单元中的主要特征节点。在GCN模块中,邻接矩阵的大小为64×64,代表了从AAA到TTT的64种可能的3-mer组合。在这个邻接矩阵中,两个3-mer被特定的间隔距离d隔开,称为在间隔d处的3-mer对。矩阵的纵轴表示该对的第一个3-mer,横轴表示第二个3-mer。邻接矩阵中的每个项(i , j)记录了3-mer ' i '在间隔d时先于3-mer ' j '出现的次数。
下面举一个简单的例子:如图2(C)所示,在d=0矩阵中,所有以AAA开头的3-mer对都放在第一行。例如,第一列(对应于对( AAA , AAA) )中的值为1,第二列(对( AAA , AAC) )中的值为2。同样,在矩阵d=1中,第二列( AAA、AAC)和第五列( AAA、ACA)中的值均为1,表明这些对在这个区间内发生一次。
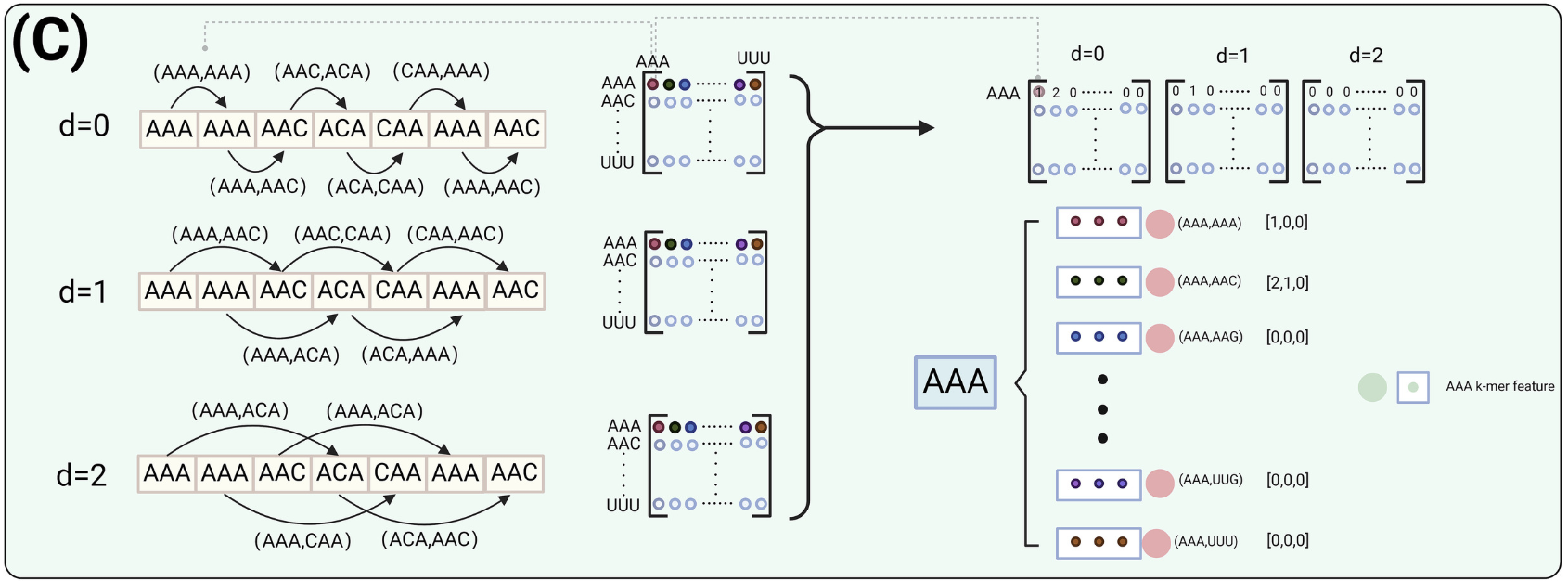
每个3-mer单元包含64个初级特征节点。主要特征节点捕获了不同间隔距离d下3-mers之间的连通性,反映了在指定距离下两个3-mers出现的频率。对于每个3-mer节点,我们考虑在区间距离d=0,d=1和d=2下的连通性。区间距离的选择是基于既要捕获lncRNA序列的结构复杂性,又要捕获其功能元件的多样性。在本研究中,3-mer单元被用作最小的图构建块,序列最初被划分为3-mer片段,如图2(C)所示。当d=0时,相邻3-mer对之间的关系被捕获。类似地,d=1和d=2捕获间隔分别为1和2的3-mer对之间的关系。如果选择d=4,两个3-mer块之间的距离将超过原始的k=3分割,从而产生间隙,破坏特征的连续性和紧密性。
数字64是指以当前3-mer开头的所有64对可能的3-mer对。例如,对于图2(C)中的AAA单元,第二个主要特征节点的值,记为(V_0、V_1、V_2),对应于邻接矩阵中3-mer对( AAA、AAC)在距离d=0,d=1和d=2处的值,其值为[ 2、1、0 ]。该编码用于为每个3-mer单元内的每个主要特征节点分配值。由邻接矩阵衍生的多尺度编码提供了lncRNA序列的鲁棒性表示,具有若干优点。首先,它捕获了跨不同区间距离的序列结构的全面视图,使模型能够理解局部和远程的3-mer关系,这在不同距离的模式都很重要的生物背景中至关重要。这种编码通过关注特定的区间,降低了维度,过滤掉了不相关的细节,突出了最相关的信息。这不仅增强了模型对细微序列模式的识别能力,而且使其对微小的序列变化不敏感,从而提高了模型的泛化能力。此外,该方法具有很高的可解释性,使研究人员能够识别哪些3-mer对和距离对预测至关重要。总体而言,这种编码方式将详细的信息表示与有效的模型性能和可解释性相结合,使其成为lncRNA序列分析的有力工具。
SAGEConv
SAGEConv (采样和聚合卷积) 是一种特殊的图卷积操作,旨在通过聚合关于其邻居的信息来学习每个节点的表示。GraphSAGE的核心思想是使用不同的聚合函数,如平均、最大池化或长短期记忆网络(LSTM)来收集周围节点的特征信息,并更新当前节点的特征表示。
在SAGEConv中,计算图(V、E)中每个节点ν的新特征表示如下:

在SAGEConv在EVlncRNA-net中的实现中,我们特别使用了平均聚合。其原因在于,平均聚合有效地捕获了节点邻居的平均信息,有利于学习鲁棒、平滑的节点表示。这种方法特别适合于构造由lncRNA序列衍生的图数据。平均集合公式表示为:
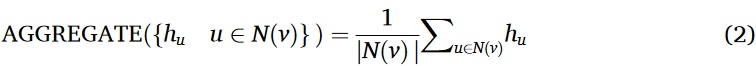
该聚合过程计算所有邻居节点的特征向量的均值,使得模型能够有效地捕获邻域信息。
在本文的方法中,使用了PyTorch Geometric (PyG )库中的SAGEConv层,该层扩展了公式( 1 ),转化为一种实用的基于图的数据工作格式。该层特别有利于处理有向或异构图,因为它允许对源节点和目标节点使用不同的节点特征。这一特性使我们能够有效地处理特征维度变化的节点。PYG的实现方式如下:
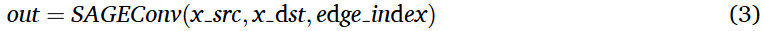
边索引(edge_index)的大小为[2 , E],其中E表示边的数量,编码节点的连接关系。
SAGEConv模块通过执行式(3)中的实际步骤,在操作上实现了式(1)中定义的计算过程。当调用SAGEConv(x_src,x)dst,edge_index)时,内部计算经历了四个不同的阶段,每个阶段都与数学表达式紧密相关。
(1)邻域识别与特征检索:维度为[2 , E]的边索引张量有利于高效地识别每个目标节点v的邻域节点N(v)。同时,该模块从源节点特征矩阵x_src中检索出所有u∈N(v)对应的特征向量hu。
(2)邻域特征聚合:聚合算子AGGREGATE(⋅)对提取的邻域特征{hu|∀u∈N(v)}进行处理,实现式(1)中规定的关键信息蒸馏步骤。
(3)特征整合:根据式(3)的公式,从目标特征矩阵x_dst直接获得的聚合邻域表示与目标节点v的内在特征hv进行级联。
(4)非线性变换:集成后的特征经过线性变换后再经过激活函数σ,最终生成更新后的节点表示hʹv,构成模块的输出。
该方法允许在我们的图的每个3-mer单元中有效地更新主特征节点和k-mer特征节点。在GCN模块中,我们使用三层SAGEConv进行节点更新。如图3所示,在每个SAGEConv层中,我们首先利用k-mer特征节点和k-mer特征节点到主特征节点的边来更新主特征节点。然后,利用更新后的主特征节点及其指向k-mer特征节点的边,更新k-mer特征节点。
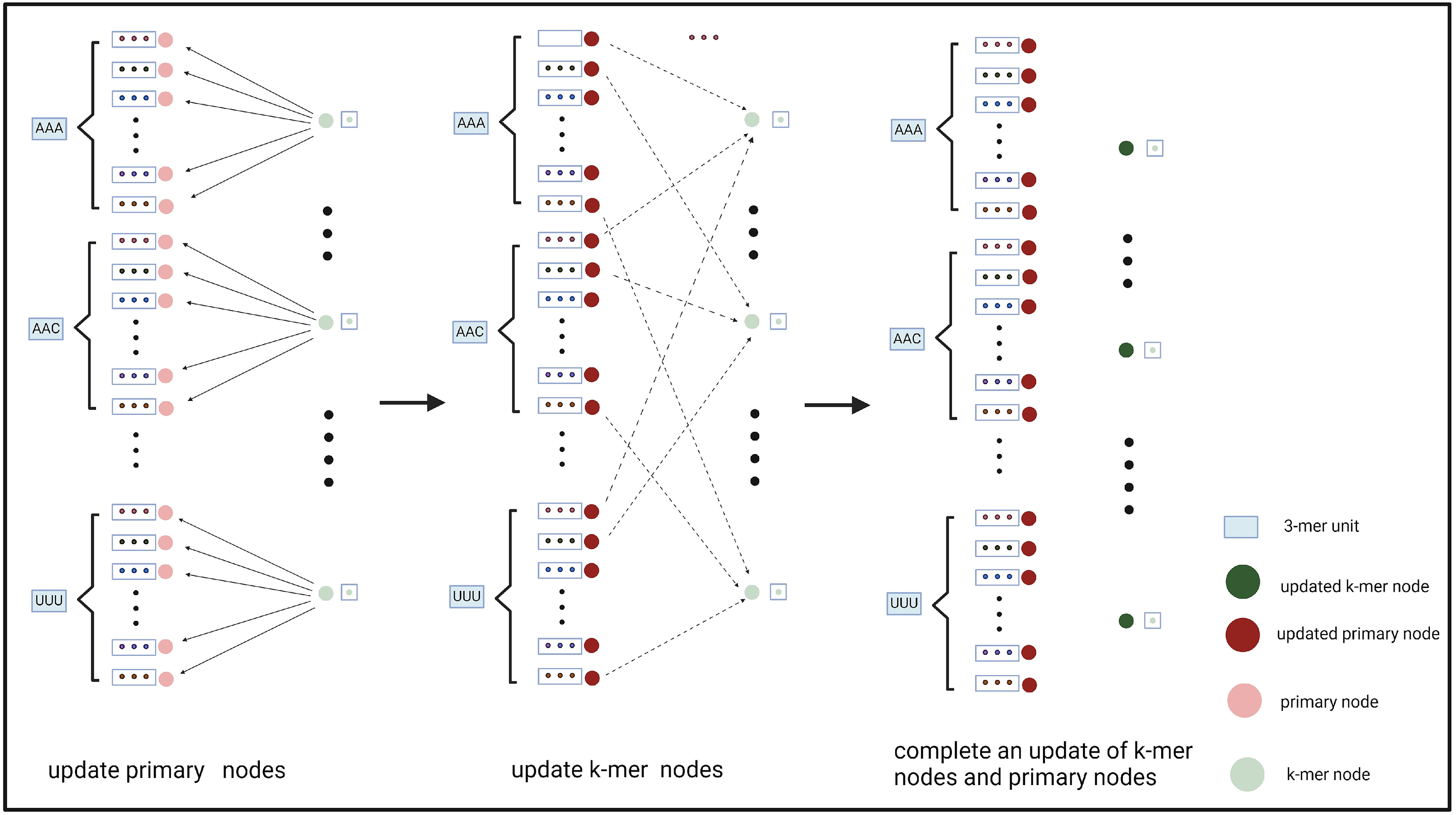
经过3层SAGEConv并进行双向更新后,将所有主要特征节点的特征进行串联。这种级联形成了由GCN衍生的最终特征表达。通过使用这种双向信息传递和特征互补的策略,我们增强了模型的表示学习能力。这种方法在捕捉序列之间错综复杂的关系和特征方面尤为有效。
GCN概述
GCN模块中,首先统计每个序列中不同的3-mers,并构建3个不同间隔距离d的邻接矩阵。每个3-mer由一个k-mer特征节点和64个主要特征节点表示。k-mer特征节点由k-mer编码得到的频率特征表征,而每个主特征节点具有由多尺度区间连接信息得到的三维特征。这些主节点捕获当前3-mer与其他所有3-mer在指定距离上的连通性。
根据建立的连接模式,将主要特征节点链接到k-mer特征节点,将序列转换为图形格式。该方法将来自k-mer特征节点的局部信息与邻接矩阵中多尺度区间连接信息提供的全局上下文信息相融合。这种综合的图构建捕获了序列内部的复杂性和相互关系,便于通过后续的GCN处理进行有效的表示学习。在这项研究中,使用SAGEConv作为图卷积算法。
CNN特征提取
one-hot编码
在生物信息学中,独热编码是一种广泛使用的特征提取方法,将生物序列转换为适合机器学习算法的格式。在此背景下,A,C,G和U这4个核苷酸碱基通常分别被编码为 [1,0,0,0],[0,1,0,0,[0,0,1,0]和 [0,0,0,1]。这种编码策略提供了一个独特的优势:它为每个核苷酸碱基或氨基酸提供了一个公平且统一的加权表示,增强了机器学习模型对生物序列数据的处理能力。
CNN概述
在CNN特征提取模块中,每个lncRNA序列都会经历一个系统的过程,以生成准确的特征表示。首先,将序列转换为一维独热编码格式。
接着,通过两个卷积层对one-hot编码序列进行处理。这些层使用多个卷积滤波器来提取序列中的重要特征,并揭示序列中的复杂模式。为了提高模型的稳定性和加速收敛,采用批归一化对跨批数据分布进行标准化处理。
随后,使用自适应最大池化在保留关键序列信息的同时降低特征的维度。最后,将处理后的特征通过一个线性层,将其细化为紧凑的表示,为后续的集成和分类任务进行优化。这种方法确保了有效地捕获和增强lncRNA序列的本质特征,以便进行进一步的分析。
模块融合
在EVlncRNA-net模型中,特征融合是必不可少的。该过程从降低从GCN模块提取的特征的维度开始,以匹配从CNN模块提取的特征的维度。如附表S1所示,我们对该步骤中的特征提取模块进行了降维实验,最终选定128维作为两个特征提取模块提出的特征的维数。
| Dimension | ACC | MCC | Sensitivity | Specificity | F1_score |
| 32 | 0.750 | 0.401 | 0.503 | 0.852 | 0.581 |
| 64 | 0.765 | 0.422 | 0.528 | 0.861 | 0.593 |
| 128 | 0.786 | 0.480 | 0.576 | 0.880 | 0.626 |
| 256 | 0.781 | 0.472 | 0.572 | 0.876 | 0.617 |
| 512 | 0.773 | 0.463 | 0.562 | 0.871 | 0.601 |
| 1024 | 0.761 | 0.445 | 0.544 | 0.866 | 0.591 |
然后将来自两个模块的精简特征拼接成一个统一的特征向量。该方法使得模型能够将来自图结构的全局信息与来自一维序列特征的局部细节信息进行融合,从而得到丰富而全面的特征表示。
融合过程从将GCN模块中的多维特征扁平化为一维向量开始。这些向量随后通过一个线性层,降低其维度以匹配类别数2,用于预测序列是否为EVlncRNA的二分类。
评估指标
评估模型的性能使用了准确率(ACC),马修斯相关系数(MCC),敏感性,特异性和F1_score:

FP(False Positives)和FN(False Negatives)表示被错误分类的正负样本。
ROC曲线下面积(AUROC)和精确回忆曲线下面积(AUPRC)也被用来评估性能。
与最先进的方法进行比较
如前所述,早期对lncRNA的研究主要集中在其鉴定上。关于预测EVlncRNAs的工作非常有限。值得注意的研究包括使用SVM算法的EVlncRNA-pred和基于深度学习的EVlncRNA-Dpred。如表8~10所示,与现有技术相比,EVlncRNA-pred表现较差。因此,本研究的重点是比较EVlncRNA-Dpred,这是一种利用深度学习的先进方法。

通过三个独立的测试比较了EVlncRNA-net和EVlncRNA-Dpred。如表8-10和图6所示,在每个数据集上,EVlncRNA-net在几乎所有指标上都优于EVlncRNA-Dpred。
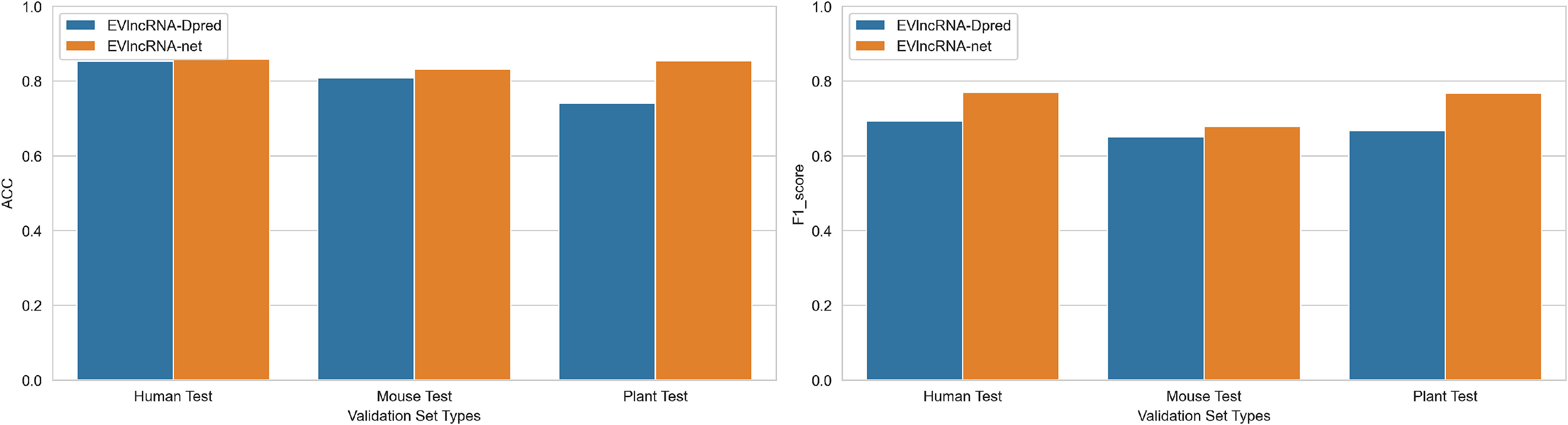
在人类数据集上,EVlncRNA-net(h)比EVlncRNA-Dpred(h)的ACC提高了0.06 %,F1分数提高了7.7 %,分别达到了0.858和0.770。
在小鼠数据集上,EVlncRNA-net(m)比EVlncRNA-Dpred(m)的准确率提高了2.3 %,F1分数提高了2.8 %,分别达到了0.831和0.678。
在植物数据集上,EVlncRNA-net(p)比EVlncRNA-Dpred(p)的准确率和F1值分别提高了11.4 %和10.0 %,达到0.854和0.767。
混合表示融合了CNN和GCN两个模块的表示,通过在有限的数据中有效地捕获细微的模式和关系,证明了混合表示在这些环境中的优势。这种组合策略有助于解决在较小数据集下经常出现的过拟合和特征表示不足等常见问题。通过同时利用局部序列信息和全局图结构,EVlncRNA-net即使从有限的数据中也能很好地提取有意义的特征。这些结果强调了该模型在识别EVlncRNA方面的卓越预测能力,证实了其作为预测工具在不同数据集规模下的稳健性和有效性。