多模态医学AI框架Pathomic Fusion,整合了组织病理学与基因组的特征
小罗碎碎念
在医学AI领域,癌症的精准诊断与预后预测一直是关键研究方向。
这篇文章提出了Pathomic Fusion这一创新框架,致力于解决现有方法的局限。

传统上,癌症诊断依赖组织学与基因组数据,但组织学分析主观易变,基因组分析难以精准区分肿瘤与正常细胞特征,且多数深度学习方法仅基于单一数据模态,未充分利用多模态数据互补信息。
文章详细阐述了Pathomic Fusion框架的构建与实现。
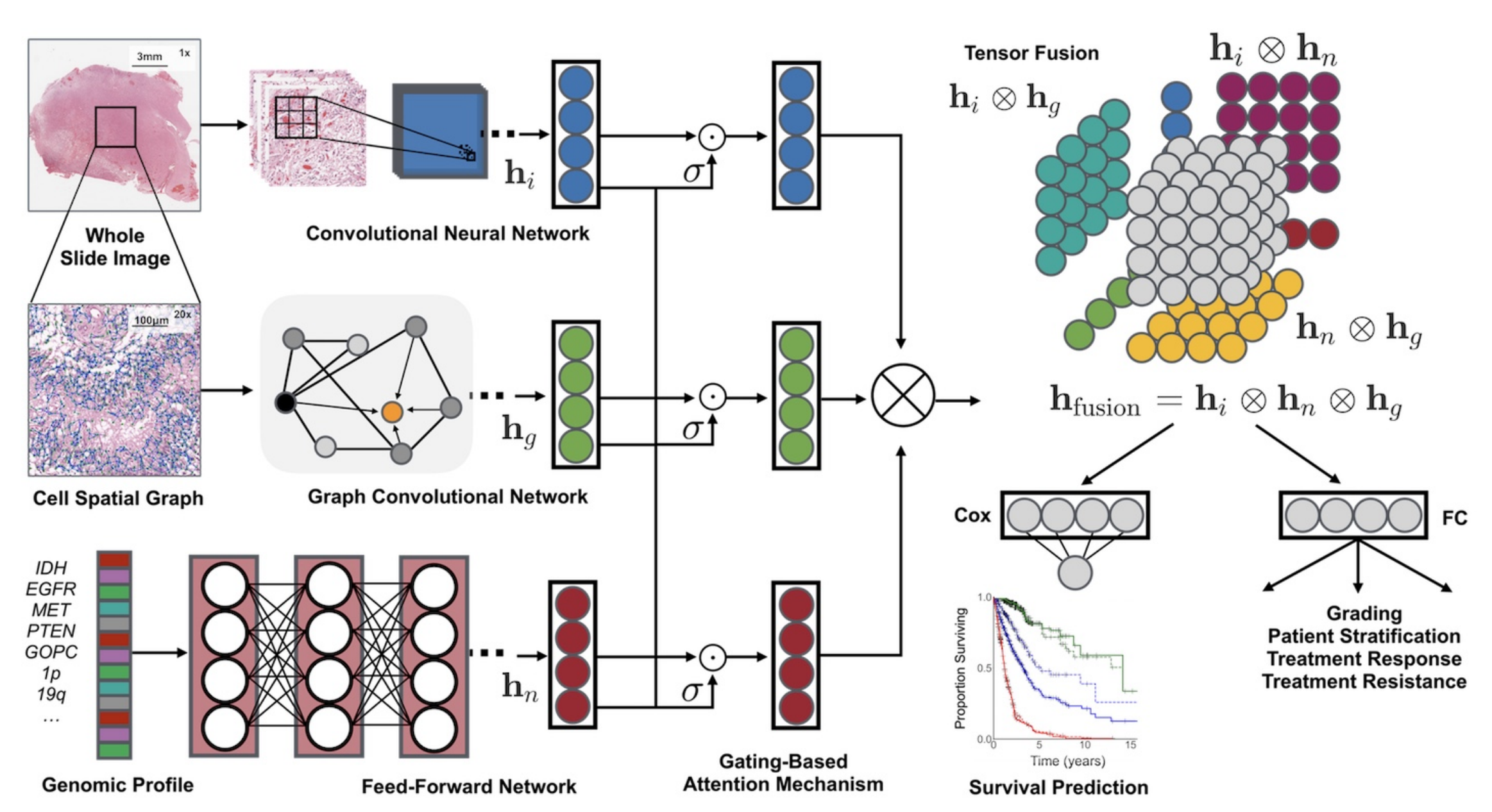
它融合了组织学图像、细胞图和基因组特征,利用卷积神经网络(CNN)和图卷积神经网络(GCN)从组织学图像中提取特征,通过自归一化网络处理基因组数据。同时,采用基于门控的注意力机制和克罗内克积来控制特征表达、建模特征交互。
在实验方面,研究人员利用来自癌症基因组图谱(TCGA)的胶质瘤和透明细胞肾细胞癌数据进行15折交叉验证,结果显示该框架在生存结果预测和患者分层上优于单模态网络和传统分级方法,并且具有良好的可解释性。
对于从事医学AI研究的人员来说,这篇文章的价值不容小觑。Pathomic Fusion框架为多模态数据融合提供了新思路,其成功应用证明了整合不同数据模态能提升癌症预测的准确性和可靠性。此外,文中的方法具有可扩展性,可应用于其他癌症类型和医学问题。
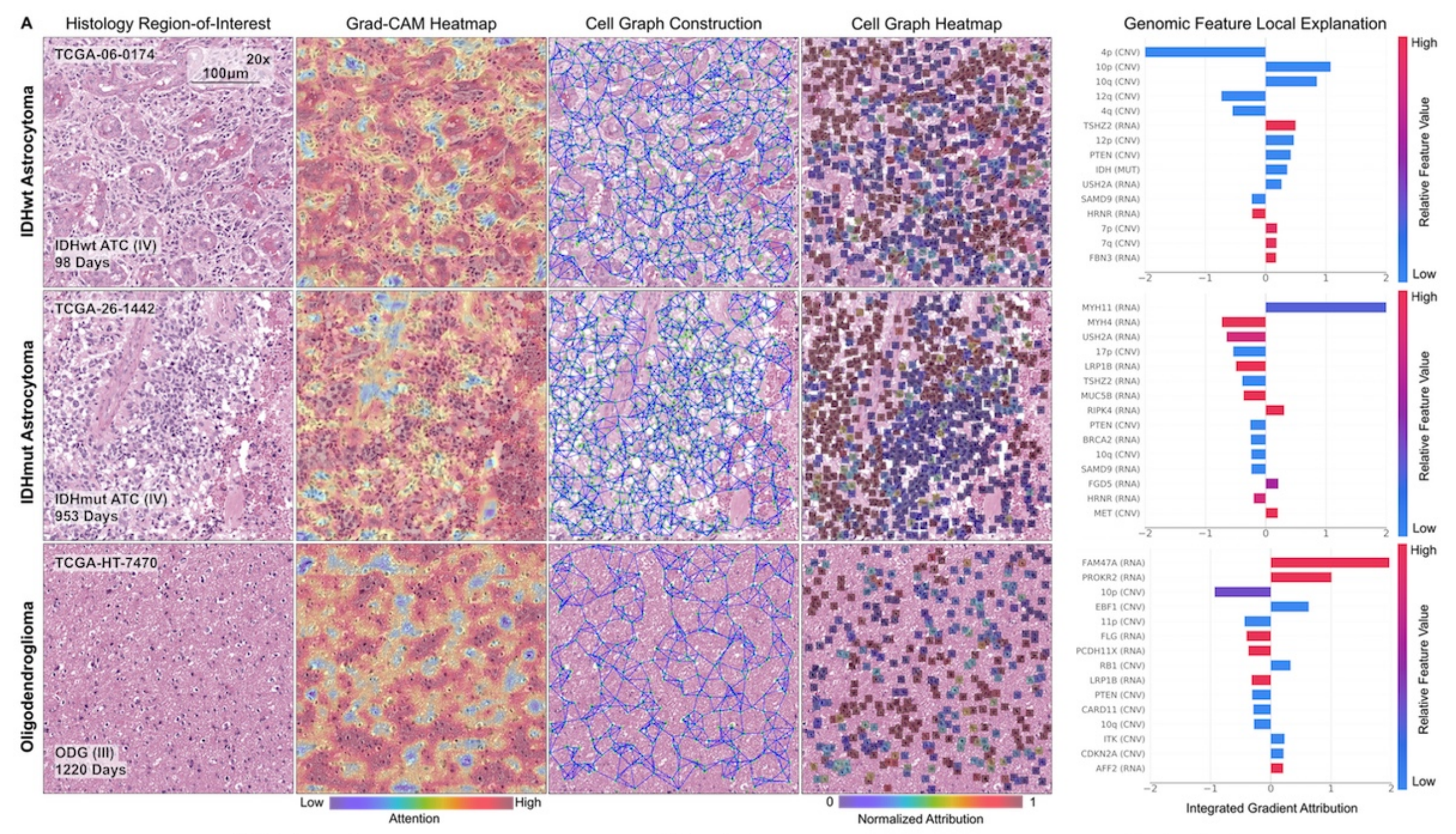
研究中的可解释性分析方法也为理解模型决策过程提供了有效手段,有助于开发更值得信赖的医学AI模型,推动医学AI在癌症诊疗领域的进一步发展。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量61,000+,交流群总成员1400+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
文章提出Pathomic Fusion框架,融合组织学和基因组特征,通过实验验证其在癌症诊断和预后预测方面的优势,为多模态生物医学数据的深度学习提供了新方法。
- 研究背景:癌症诊断、预后和治疗反应预测依赖组织学和基因组数据,但现有方法存在局限性。组织学分析主观且存在观察者间差异,基因组分析无法精准区分肿瘤与非肿瘤细胞的基因变化。多模态深度学习虽发展迅速,但生物医学领域的融合策略尚待探索。
- 研究方法
- Pathomic Fusion框架:创新性地融合组织学图像、细胞图和基因组特征。组织学特征通过CNN和GCN提取,基因组特征由自归一化网络提取,采用基于门控的注意力机制和克罗内克积构建多模态张量,实现特征交互建模。
- 实验设计:收集TCGA中胶质瘤和透明细胞肾细胞癌数据,进行15折交叉验证。对比不同模型配置和融合策略,以Cox比例风险模型为基线,使用一致性指数(c-Index)等指标评估模型性能。
- 实验结果
- 生存预测性能:Pathomic Fusion在胶质瘤和透明细胞肾细胞癌的生存预测上优于单模态网络和WHO分级范式。在胶质瘤中,c-Index达到0.826,相比WHO范式和之前的方法有显著提升。
- 患者分层能力:能更精细地分层患者生存曲线。在胶质瘤中,其数字分级与分子亚型相关,可更好地区分中高风险患者;在透明细胞肾细胞癌中,能区分不同生存时长的患者,且分级与Fuhrman分级系统相符。
- 可解释性:通过修改Grad-CAM和Integrated Gradients方法,可解释模型在生存预测中对各模态特征的使用。在胶质瘤和透明细胞肾细胞癌中,均能识别重要的基因组特征和组织学特征。
- 研究结论:Pathomic Fusion是一种有效的多模态融合框架,可用于构建客观的图像组学检测方法,实现癌症的精准诊断和预后预测。该方法具有可扩展性和可解释性,有助于发现新的生物标志物,为癌症治疗提供指导 。
二、模型架构
2-1:多模态融合框架
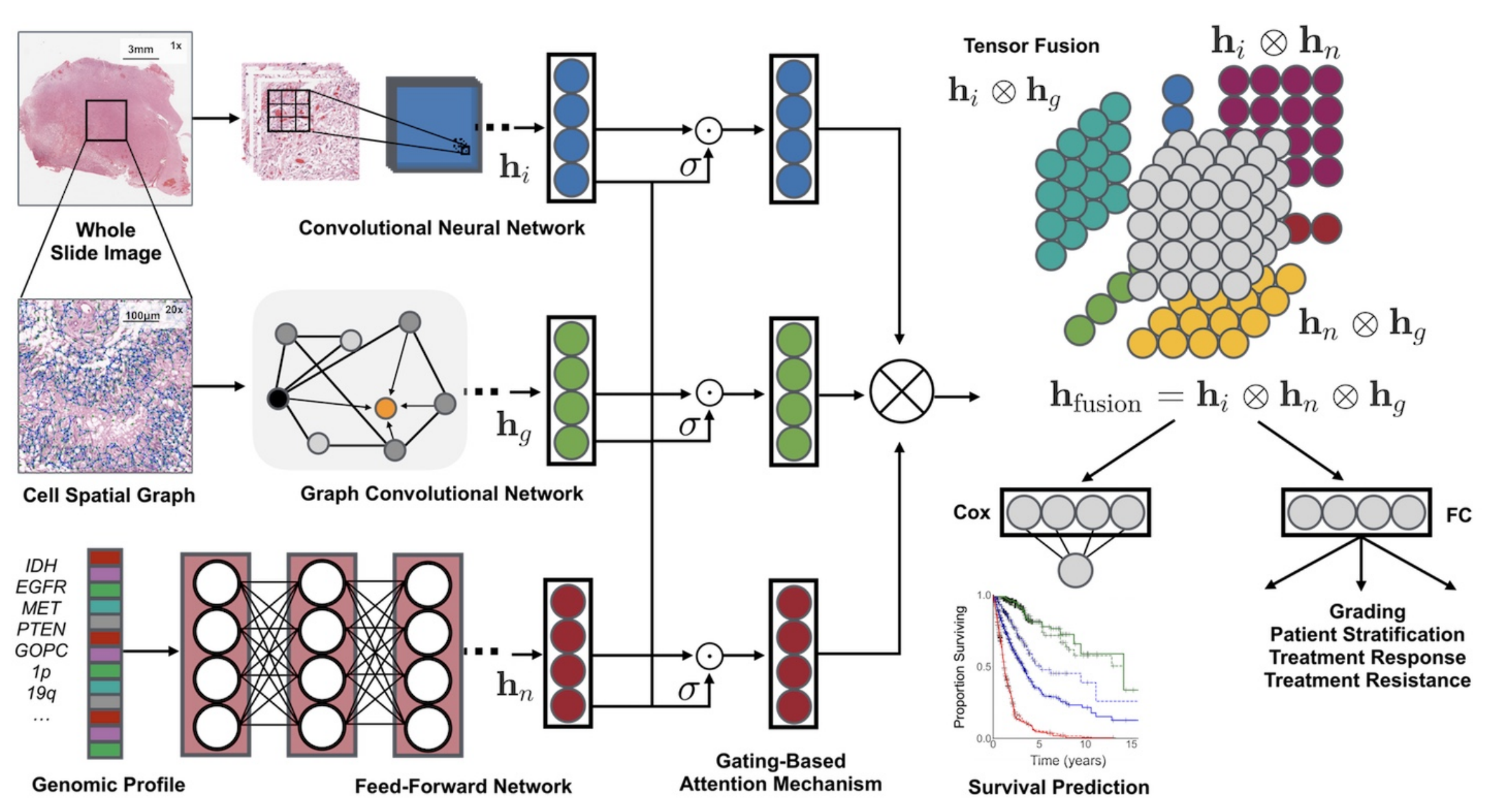
特征提取
- 组织学图像特征( h i h_i hi):从全切片图像(Whole Slide Image)选取区域,经卷积神经网络(Convolutional Neural Network, CNN)处理提取特征。可单独用CNN,或结合参数高效的图卷积网络(Graph Convolutional Network, GCN) 。
- 细胞空间图特征( h g h_g hg):对组织学图像构建细胞空间图(Cell Spatial Graph),通过图卷积网络(GCN)提取细胞形态等特征。
- 基因组特征( h n h_n hn):针对基因组图谱(Genomic Profile ,如IDH、EGFR等基因标识 ),利用前馈网络(Feed - Forward Network)提取特征。
特征处理与融合
- 门控注意力机制(Gating - Based Attention Mechanism):对各模态特征分别处理,通过门控机制( σ \sigma σ ,类似激活函数)控制各模态特征表达程度,突出重要特征 。
- 张量融合(Tensor Fusion):运用克罗内克积( ⊗ \otimes ⊗ )对处理后的不同模态特征进行两两交互建模,得到融合特征 h f u s i o n = h i ⊗ h n ⊗ h g h_{fusion}=h_i \otimes h_n \otimes h_g hfusion=hi⊗hn⊗hg 。
下游任务应用
- 生存预测(Survival Prediction):将融合特征输入Cox层,依据Cox比例风险模型预测患者生存情况,输出生存曲线。
- 分级及其他应用:经全连接层(FC)进行分级,用于患者分层、治疗反应预测、治疗抵抗评估等。
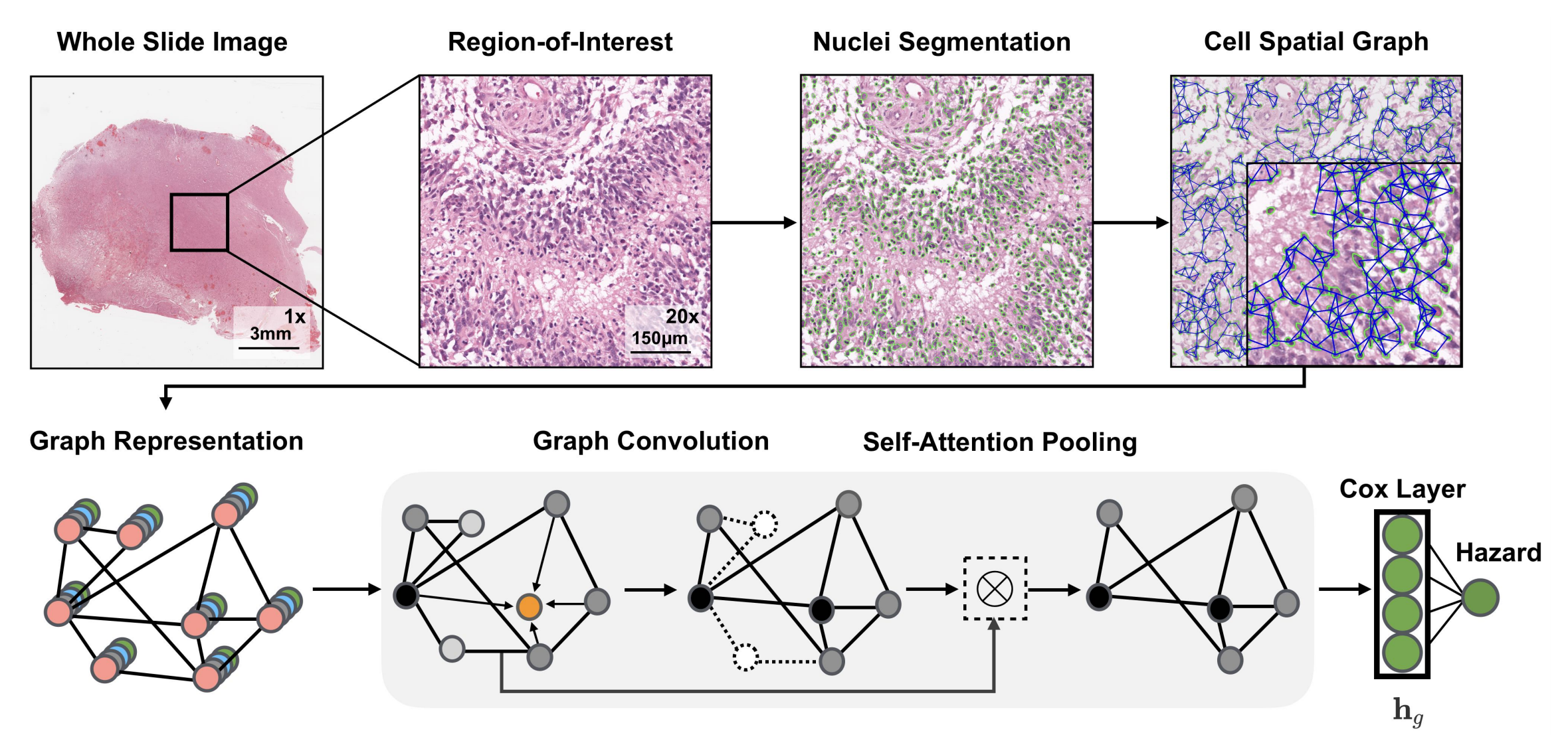
2-2:图卷积网络架构
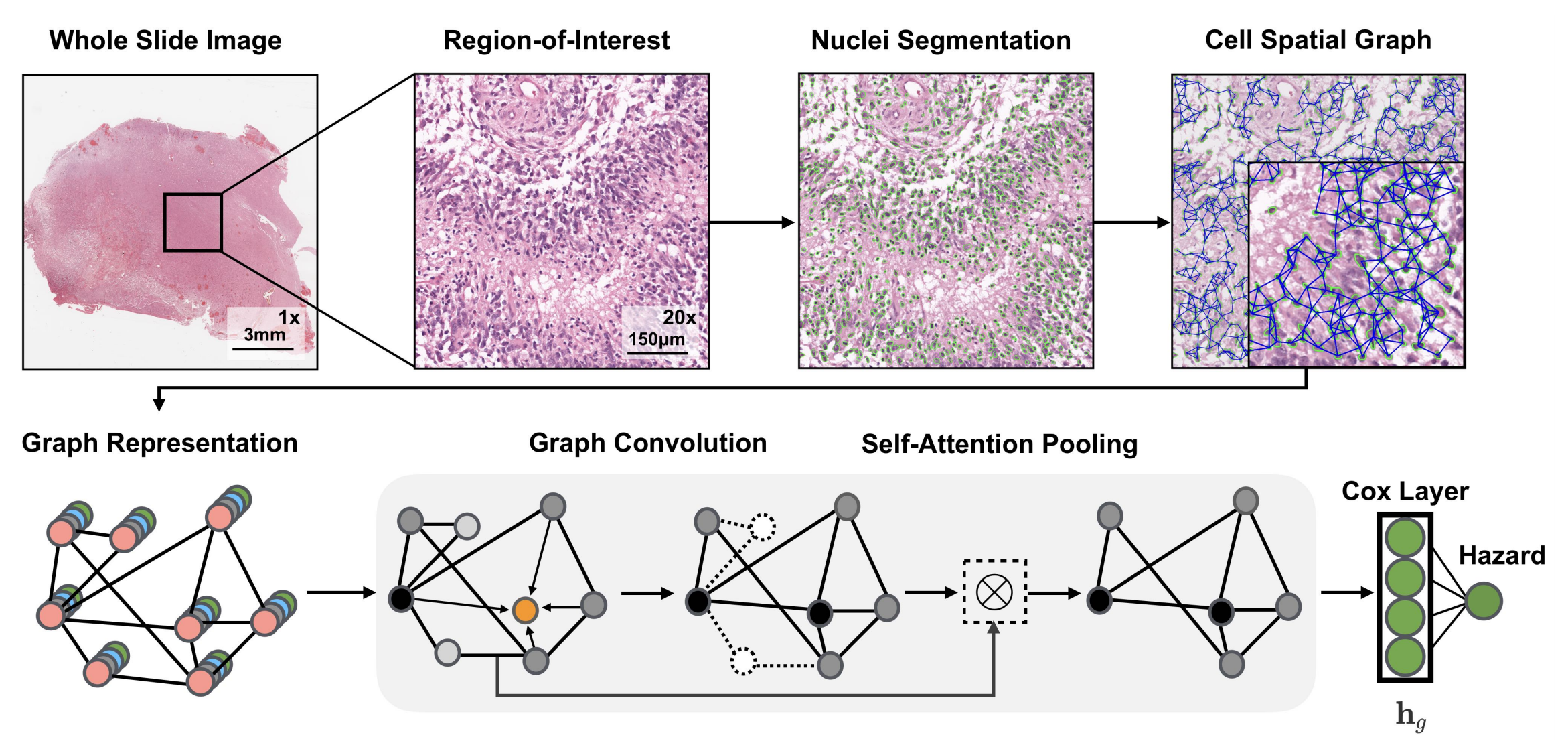
- 全切片图像(Whole Slide Image)处理:获取全切片组织学图像,选取其中感兴趣区域(Region - of - Interest ) ,对细胞进行分割(Nuclei Segmentation),构建细胞空间图(Cell Spatial Graph) ,将细胞视为图中的节点。
- 图表示(Graph Representation):基于深度学习的细胞核分割算法分离细胞,用K近邻(KNN)确定细胞间连接 。细胞特征通过手工设计特征和对比预测编码学习的深度特征初始化。
- 图卷积(Graph Convolution):采用GraphSAGE架构的聚合和组合函数,对图节点特征进行卷积操作,更新节点特征。
- 自注意力池化(Self - Attention Pooling):借鉴SAGEPool的节点掩码和分层池化策略,通过自注意力机制对图节点进行池化操作,突出重要节点 。
- Cox层(Cox Layer):将处理后的特征输入Cox层,计算风险值(Hazard) ,用于生存分析等任务。
三、生存分析
生存分析是对某一事件发生时间进行建模的任务。
在实际情况中,并非所有事件的结果都能完整观测到,这种情况被称为删失(censored)。
以癌症生存结果预测为例,患者死亡是未删失事件,而患者存活或最后一次已知随访则属于删失事件,此时最后一次已知接触日期就作为生存时间的下限。
设 T T T 为连续随机变量,表示患者的生存时间,生存函数 S ( t ) = P ( T ≥ t 0 ) S(t)=P(T \geq t_0) S(t)=P(T≥t0),即表示患者生存时间超过 t 0 t_0 t0 的概率。
3-1:风险函数
风险函数 λ ( t ) \lambda(t) λ(t) 定义为在 t t t 时刻( t 0 t_0 t0 之后)事件瞬间发生的概率,其数学表达式为:
λ ( t ) = lim Δ t → 0 P ( t ≤ T ≤ t + Δ t ∣ T ≥ t ) Δ t \lambda(t)=\lim_{\Delta t \to 0} \frac{P(t \leq T \leq t + \Delta t | T \geq t)}{\Delta t} λ(t)=Δt→0limΔtP(t≤T≤t+Δt∣T≥t)
这个公式的含义是,在已经存活到 t t t 时刻的条件下,在极短时间间隔 Δ t \Delta t Δt 内事件发生的概率与 Δ t \Delta t Δt 的比值,当 Δ t \Delta t Δt 趋于 0 时的极限 。
生存函数 S ( t ) S(t) S(t) 与风险函数 λ ( t ) \lambda(t) λ(t) 存在如下关系:
S ( t ) = exp ( − ∫ 0 t λ ( x ) d x ) S(t)=\exp\left(-\int_{0}^{t} \lambda(x)dx\right) S(t)=exp(−∫0tλ(x)dx)
推导过程
从风险函数定义出发,对 S ( t ) S(t) S(t) 关于 t t t 求导,根据概率的基本性质有:
d S ( t ) d t = − λ ( t ) S ( t ) \frac{dS(t)}{dt}=- \lambda(t)S(t) dtdS(t)=−λ(t)S(t)
这是一个可分离变量的微分方程,将其变形为 d S ( t ) S ( t ) = − λ ( t ) d t \frac{dS(t)}{S(t)}=-\lambda(t)dt S(t)dS(t)=−λ(t)dt ,然后对两边同时积分:
∫ S ( 0 ) S ( t ) d S S = − ∫ 0 t λ ( x ) d x \int_{S(0)}^{S(t)} \frac{dS}{S}=-\int_{0}^{t} \lambda(x)dx ∫S(0)S(t)SdS=−∫0tλ(x)dx
已知 S ( 0 ) = 1 S(0) = 1 S(0)=1 ,对左边积分可得 ln S ( t ) − ln 1 = ln S ( t ) \ln S(t)-\ln 1 = \ln S(t) lnS(t)−ln1=lnS(t) ,所以得到 S ( t ) = exp ( − ∫ 0 t λ ( x ) d x ) S(t)=\exp\left(-\int_{0}^{t} \lambda(x)dx\right) S(t)=exp(−∫0tλ(x)dx) 。
3-2:Cox比例风险模型
Cox比例风险模型是估计风险函数最常用的半参数方法。
它假设风险函数可以参数化为指数线性函数:
λ ( t ∣ x ) = λ 0 ( t ) e β x \lambda(t|x)=\lambda_0(t)e^{\beta x} λ(t∣x)=λ0(t)eβx
其中, λ 0 ( t ) \lambda_0(t) λ0(t) 是基线风险函数,描述了事件在时间 t t t 上的基础风险,它不依赖于协变量 x x x ; β \beta β 是模型参数向量, x x x 是患者的协变量(如基因特征、临床指标等), β x \beta x βx 描述了风险如何随着协变量 x x x 的变化而变化 。
在原始模型中,基线风险 λ 0 ( t ) \lambda_0(t) λ0(t) 的具体形式未被指定,这使得直接估计 β \beta β 变得困难。不过,可以通过推导Cox部分对数似然函数来估计 β \beta β 。
Cox部分对数似然函数 l ( β , X ) l(\beta, X) l(β,X) 为:
l ( β , X ) = − ∑ i ∈ U ( X i β − log ∑ j ∈ R i e X j β ) l(\beta, X)=-\sum_{i \in U} \left( X_i \beta - \log \sum_{j \in R_i} e^{X_j \beta} \right) l(β,X)=−i∈U∑ Xiβ−logj∈Ri∑eXjβ
其中, U U U 是未删失患者的集合, R i R_i Ri 是死亡时间或最后随访时间晚于患者 i i i 的患者集合 。
对 l ( β , X ) l(\beta, X) l(β,X) 关于 β \beta β 求偏导:
∂ l ( β , X ) ∂ β = ∑ i ∈ U ( X i − ∑ j ∈ R i X j e X j β ∑ j ∈ R i e X j β ) \frac{\partial l(\beta, X)}{\partial \beta}=\sum_{i \in U} \left( X_i - \frac{\sum_{j \in R_i} X_j e^{X_j \beta}}{\sum_{j \in R_i} e^{X_j \beta}} \right) ∂β∂l(β,X)=i∈U∑(Xi−∑j∈RieXjβ∑j∈RiXjeXjβ)
为了更方便地表示,令 δ ( i ) \delta(i) δ(i) 为指示变量,当患者 i i i 未删失时 δ ( i ) = 1 \delta(i) = 1 δ(i)=1 ,删失时 δ ( i ) = 0 \delta(i) = 0 δ(i)=0 ,则偏导可写成:
∂ l ( β , X ) ∂ β = ∑ i ∈ U ( δ ( i ) X i − ∑ j ∈ R i δ ( j ) X j e X j β ∑ j ∈ R i e X j β ) \frac{\partial l(\beta, X)}{\partial \beta}=\sum_{i \in U} \left( \delta(i)X_i - \frac{\sum_{j \in R_i} \delta(j)X_j e^{X_j \beta}}{\sum_{j \in R_i} e^{X_j \beta}} \right) ∂β∂l(β,X)=i∈U∑(δ(i)Xi−∑j∈RieXjβ∑j∈Riδ(j)XjeXjβ)
可以使用迭代优化算法,如牛顿 - 拉夫森(Newton - Raphson)算法或随机梯度下降(Stochastic Gradient Descent)算法来估计 β \beta β 。
3-3:深度学习在生存分析中的应用及模型评估
训练用于生存分析的深度网络时,隐藏层特征作为Cox模型协变量,部分对数似然函数导数作为反向传播误差 。
评估生存分析网络性能用一致性指数(c - Index) ,衡量预测风险得分与患者真实生存时间排序一致性 。为展示Pathomic Fusion性能,与其他模型对比用c - Index 。
临床实践基线是在Cox比例风险模型中用真实分子亚型作为协变量 。通过Log Rank检验计算P值,评估不同风险分层 ,如胶质瘤的低、中、高风险(33 - 66 - 100百分位数),透明细胞肾细胞癌(CCRCC)的25 - 50 - 75 - 100百分位数风险分层 。
四、方法细节
4-1:基因组和转录组特征的纳入标准
在对合并的TCGA - GBMLGG和TCGA - KIRC项目进行分析时,分别使用了320个和357个基因组特征。
基因组特征包括突变(如IDH1基因的突变状态二元指示,0/1 )和拷贝数变异(CNV )(如基因和染色体区域的扩增/缺失拷贝 )。TCGA中拷贝数变异的测量使用Affymetrix SNP 6.0芯片来识别基因组区域的重复拷贝,最终输出为片段平均值(扩增区域为正值,缺失区域为负值 )。
对于TCGA - GBMLGG,本分析中使用的突变和CNV数据是从Mobadersany等人[29]使用的同一组基因组特征中整理而来。整理的基因包括EGFR、MDM4、MGMT、MYC和BRAF,这些基因与血管生成、细胞凋亡、细胞生长和分化等致癌过程有关。
对于TCGA - KIRC,使用了扩增/缺失最多的基因(所有扩增或缺失大于7%的CNV ),得到117个CNV特征。对于这两个项目,均纳入了RNA - Seq表达数据,其以mRNA转录本的定量总体丰度来衡量。
通过cBioPortal,为两个项目均选择了前240个差异表达基因[65]。由于基因组特征之间不存在任何明确的空间或时间依赖性,因此直接输入特征。
4-2:TCGA - GBMLGG中的数据缺失和对齐
为与先前最先进的研究进行比较,使用了[29]附录中现有的整理后的TCGA - GBMLGG数据,这需要仔细处理多模态数据中的缺失值。
对于每位患者,使用来自诊断切片的1 - 3个20倍放大、1024 × 1024大小(0.5 µ /像素 )的组织学感兴趣区域(ROI ),以及320个基因组特征。在769名患者中,72名患者缺失分子亚型(IDH突变和1p19q共缺失 )信息,33名患者缺失组织学亚型和分级标签,256名患者缺失mRNA - Seq数据(图6 )。
由于部分患者有多个来自诊断切片的ROI,在交叉验证中每个图像被视为一个单独的数据点,同时复制基因组和真实标签信息。使用与[29]附录相同的训练 - 测试划分进行15折蒙特卡罗交叉验证,该划分按TCGA ID随机生成,80%用于训练,20%用于测试。
由于数据缺失,根据任务(生存预测与分级分类 )和使用的模态组合(组织学、基因组、组织学 + 基因组 ),从训练划分中选取不同子集来训练单模态和多模态网络。
在对存在缺失数据的交叉验证测试划分上验证模型时,对测试划分进行标准化处理,以排除所有模型中的缺失数据(图5中的中心重叠部分 )。
在处理透明细胞肾细胞癌(CCRCC )数据时,数据缺失不是问题,所有单模态和多模态网络均使用相同的训练 - 测试划分进行15折交叉验证。
4-3:网络架构
本研究使用三种不同的网络架构来处理三种模态的数据:
- 1)用于组织学图像的带批量归一化的VGG19卷积神经网络(CNN );
- 2)用于细胞空间图的图卷积网络(GCN );
- 3)用于分子特征谱的前馈自归一化网络。
VGG19网络由16个卷积层、3个全连接层和5个最大池化层组成,输入图像大小为512 × 512。在前两个全连接层(大小为1024 )后应用丢弃率为0.25的Dropout,在最后一个隐藏层(大小为32 )后应用较低丢弃率(p = 0.05 )的Dropout。
GCN由3个GraphSAGE层和自注意力池化层组成,隐藏层维度为128,随后是两个大小分别为128和32的线性层。
基因组自归一化网络(Genomic SNN )由4个连续的全连接层模块组成,维度分别为[64, 48, 32, 32],采用ELU激活函数和Alpha Dropout。
对于生存结果预测,所有网络使用Sigmoid函数激活,输出缩放至 - 3到3之间。对于分级分类,所有网络使用Log Softmax激活,以计算三个WHO分级各自的得分。
多模态网络架构由两个部分组成:
- 1)基于门控的模态注意力;
- 2)通过克罗内克积进行融合。
每种模态通过三个线性层进行门控,第二个线性层用于计算注意力分数。
对于生存结果预测,使用基因组模态对图像和图模态进行门控;对于分级分类,使用组织学图像模态对基因组和图模态进行门控。对门控后的单模态特征表示进行额外降维,以减小三模态网络中克罗内克积特征空间的输出大小。
在三模态网络中,对于生存结果预测,基因组模态的第一和第三个线性层有32个隐藏单元以保持特征图维度,图像和图模态的线性层有16个隐藏单元,以便将特征表示转换为较低维度。
对于分级分类,保持组织学图像模态的特征维度,降低图和基因组模态的维度。在任何任务的双模态网络中均不进行特征维度降低。
对于特征融合,计算每种模态各自单模态特征表示的克罗内克积,对于CNN⊗SNN、GCN⊗SNN和CNN⊗GCN⊗SNN,分别创建大小为[33 × 33]、[33 × 33]、[33 × 17 × 17]的特征图。
为使用未扰动的单模态特征,在计算克罗内克积之前向每个特征向量添加1。在门控和计算多模态张量后插入丢弃率为(p = 0.25 )的Dropout层。
4-4:实验细节
Pathomic Fusion使用PyTorch 1.5.0、PyTorch Geometric 1.5.0、Captum 0.2.0和Lifelines 0.24.6构建。
用于构建细胞图的节点特征通过以下方式计算:
- 1)分割每个细胞核;
- 2)使用OpenCV 4.2.0中的轮廓特征工具箱;
- 3)纹理特征工具箱;
- 4)使用对比预测编码的自监督深度特征;
- 5)使用PyFlann 1.6.14进行图构建。
实验使用的资源包括本地工作站上的12块英伟达GeForce RTX 2080 Ti显卡,以及谷歌云平台上的2块英伟达Tesla V100显卡。
组织学CNN使用来自ImageNet的预训练权重进行初始化,随后使用0.0005的低学习率和8的批量大小对网络进行微调。通过随机裁剪512 × 512大小图像、颜色抖动以及随机垂直和水平翻转进行数据增强。
组织学GCN和基因组SNN使用Klambeur等人[58]提出的自归一化权重进行初始化,分别使用0.002的学习率,以及32和64的批量大小进行训练。对于基因组SNN,还使用了超参数值为3e - 4的轻度L1正则化以强制特征稀疏性。所有网络使用Adam优化器、丢弃率p = 0.25以及线性衰减学习率调度器,训练轮次相同。
在训练组织学CNN后,对于每个1024 × 1024的组织学ROI,从9个重叠的512 × 512图像块中提取[32 × 1]的嵌入向量,将其与各自的细胞图和基因组特征输入配对,作为Pathomic Fusion的输入。
对于组织学GCN和基因组SNN,首先按照上述训练细节训练各自的单模态网络,然后在冻结单模态网络模块的情况下,使用0.0001的学习率和Adam求解器训练多模态网络的最后线性层。
在第5轮训练时,解冻基因组和图网络,然后使用0.0001的学习率、Adam求解器和线性衰减学习率调度器再训练网络25轮。
4-5:评估细节
在15折交叉验证的测试划分上评估每个单模态和多模态网络预测的风险值和分级得分。
为了在TCGA - GBMLGG上基于CNN进行生存结果预测时使用整个1024 × 1024的组织学图像,与先前工作类似,计算属于每位患者的所有组织学ROI中9个重叠的512 × 512图像裁剪块的风险预测平均值。
为绘制卡普兰 - 迈耶(Kaplan - Meier)曲线,汇集15折交叉验证中所有测试划分的预测风险值,并根据生存时间进行绘制。为创建散点图(Swarm plots ),在汇集之前对每个划分中的预测风险值进行z - 分数标准化,以便在可视化中低风险与中风险的得分范围相似。
对于TCGA - GBMLGG上的分级分类,使用重叠的512 × 512图像块的最大softmax激活得分来确定类别。对于在CCRCC上基于CNN的生存结果预测,类似地计算每位患者512 × 512组织学ROI的风险预测平均值。
五、项目梳理
注意,这只是初步梳理,并不是详细的复现的教程。
Pathomic Fusion 是一个整合组织病理学图像和基因组特征的多模态融合框架,用于癌症诊断和预后预测。其核心创新点在于使用注意力门控(Attention Gating)和Tensor融合技术,支持卷积神经网络(CNN)、图卷积网络(GCN)或其组合处理数据。
5-1:环境配置
系统要求
• 操作系统:Linux(推荐 Ubuntu 18.04+)
• 硬件:NVIDIA GPU(如 RTX 2080 Ti 或 V100)
• 依赖项:
◦ CUDA 10.1 + cuDNN 7.5
◦ PyTorch ≥1.1.0
◦ torch_geometric=1.3.0
安装步骤
# 安装 PyTorch
conda install pytorch==1.7.1 torchvision==0.8.2 cudatoolkit=10.1 -c pytorch# 安装 torch-geometric
pip install torch-scatter -f https://pytorch-geometric.com/whl/torch-1.7.0+cu101.html
pip install torch-sparse -f https://pytorch-geometric.com/whl/torch-1.7.0+cu101.html
pip install torch-geometric==1.7.0
5-2:数据准备
目录结构
按以下结构组织数据:
data/
└─ PROJECT_NAME/├─ Images/ # 组织病理图像(.png)├─ Graphs/ # 图数据(.pkl)└─ Genomics/ # 基因组数据(.csv)
数据对齐
• 运行 make_splits.py 生成跨模态数据对齐文件:
python make_splits.py --project PROJECT_NAME --data_root ./data/
• 生成 splits.pkl 文件记录数据路径和划分。
5-3:代码结构解析
关键文件说明
• train_cv.py: 执行交叉验证训练。
• test_cv.py: 在测试集评估模型。
• networks.py: 定义单模态和多模态网络模型。
• fusion.py: 多模态融合机制实现。
• data_loaders.py: 数据加载器,支持多模态输入。
5-4:模型训练与评估
单模态训练(示例:组织病理图像)
python train_cv.py \--exp_name survival_prediction \--dataroot ./data/TCGA_GBMLGG/ \--task surv \--mode A \--model_name CNN_A \--batch_size 64 \--lr 0.002 \--gpu_ids 0
• mode A: 指定使用图像模态。
• task surv: 生存预测任务。
多模态融合训练(图像+基因组)
python train_cv.py \--task grad \--mode AB \--model_name Fusion_AB \--fusion_type tensor \--lr 0.001 \--niter_decay 100
• fusion_type tensor: 使用Tensor融合策略。
模型测试
python test_cv.py \--checkpoints_dir ./checkpoints/TCGA_GBMLGG/ \--model Fusion_AB \--phase test
5-5:复现论文结果
下载预处理数据
访问 Google Drive 下载 TCGA-GBMLGG 和 TCGA-KIRC 数据集。
运行基线模型
python run_cox_baselines.py \--omics_path ./data/genomic_data.csv \--survival_path ./data/survival_labels.csv
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!