代谢组数据分析(二十四):基于tidymass包从质谱原始数据到代谢物注释结果的实践指南
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!
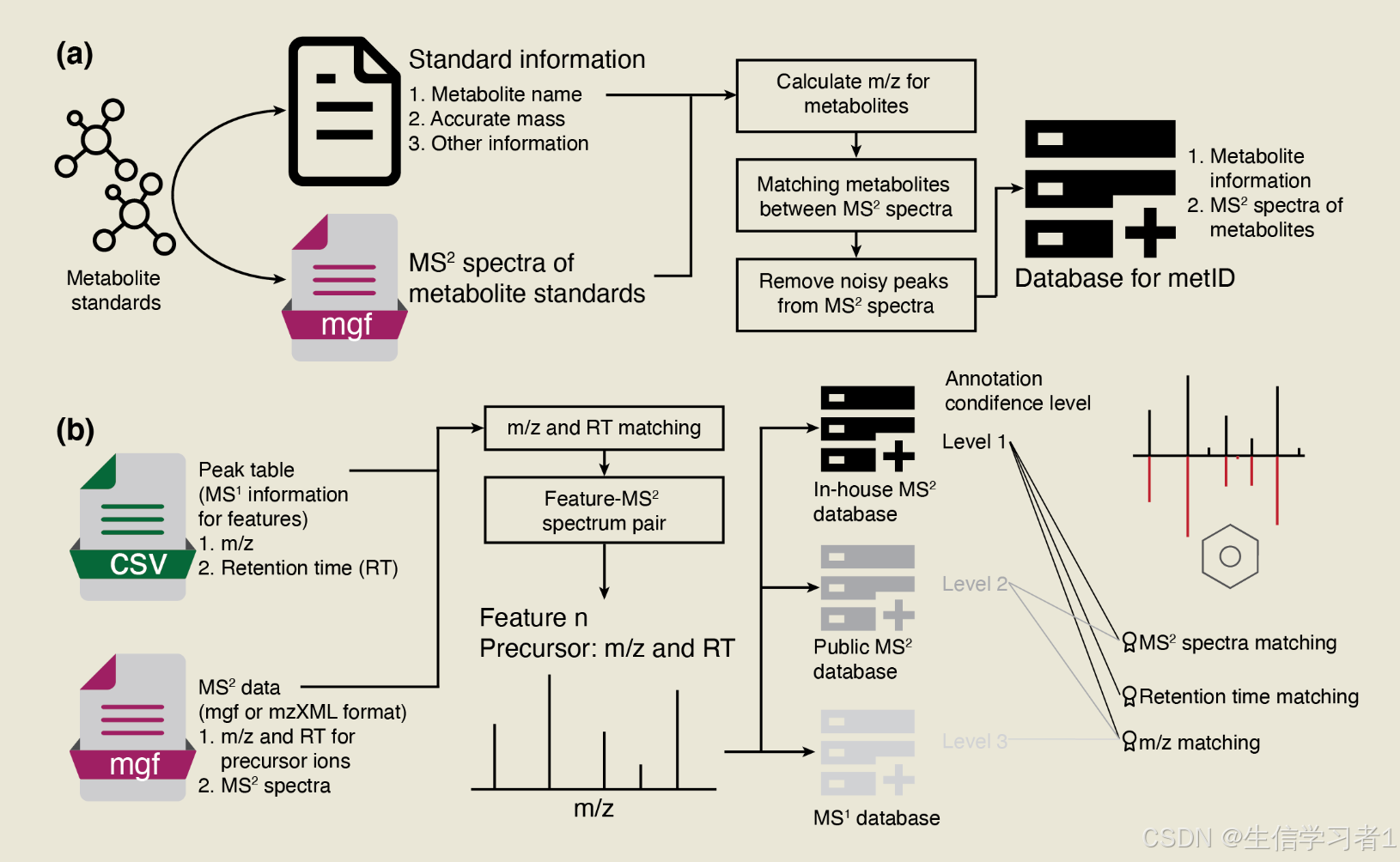
文章目录
- 介绍
- 加载R包
- 数据准备
- 原始数据处理
- 导入massDataset数据对象
- 交互图
- 数据探索
- 更新样本表格信息
- 峰分布情况
- 缺失值情况
- 数据清洗
- 数据质量评估
- 去除噪声代谢特征
- 过滤立群样本
- 填补缺失值
- 数据标准化和整合
- 预处理后评估
- 代谢物注释
- 增加MS2图谱到数据对象
- 数据库1注释
- 数据库2注释
- 数据库3注释
- 结果
- 统计分析
- 剔除无注释代谢物
- 追踪数据对象的相关处理信息
- 剔除冗余代谢物
- 差异代谢物
- 倍数变化
- 差异P值
- 火山图
- 输出结果
- 总结
- 参考
- 系统信息
介绍
本教程旨在帮助用户通过 R 语言中的 tidymass 包进行代谢组学数据分析。代谢组学是一种研究生物体内所有代谢物及其变化规律的科学,广泛应用于疾病诊断、药物研发等领域。tidymass 是一个基于 R 的开源包,专为液相色谱-质谱(LC-MS)数据分析设计,提供了一套完整的数据处理、注释和分析框架。
1. 数据准备
用户需要准备以下三种数据:
- MS1 数据:包含样本的 mzXML 文件,用于一级质谱数据。
- MS2 数据:包含 QC 样本的 mgf 文件,用于二级质谱数据。
- 样本信息表:以 Excel 格式存储,包含样本的元数据信息。
2. 数据处理
数据处理是代谢组学分析的关键步骤。tidymass